Modern Oxidation Methods Jan-Erling Backvall Title: Modern Oxidation Methods Author: Jan-Erling Backvall This is an exact replica of a book published in 2004. The book reprint was manually improved by a team of professionals, as opposed to automatic/OCR processes used by some companies. However, the book may still have imperfections such as missing pages, poor pictures, errant marks, etc. that were a part of the original text. We appreciate your understanding of the imperfections which can not be improved, and hope you will enjoy reading this book. 1.2 Environmentally Friendly Terminal Oxidants
1.2.2 Hypochlorite
Apart from oxygen and hydrogen peroxide, bleach is the simplest and cheapest oxi-dant that can be used in industry without problems. In the past this oxidant has onlybeen applied in the presence of osmium complexes in two patents in the early 1970sfor the oxidation of fatty acids [20]. In 2003 the first general dihydroxylation proce-dure of various olefins in the presence of sodium hypochlorite as the reoxidant wasdescribed by us [21]. Using a-methylstyrene as a model compound, 100 % conversionand 98 % yield of the desired 1,2-diol were obtained (Scheme 1.5). Scheme 1.5 Osmium-catalyzed dihydroxylation of a-methylstyrene using sodium hypochlorite
Interestingly, the yield of 2-phenyl-1,2-propanediol after 1 h was significantly
higher using hypochlorite compared with literature protocols using NMO (90 %) [22]or K3[Fe(CN)6] (90 %) at this temperature. The turnover frequency was 242 h–1,which is a reasonable level [23]. Under the conditions shown in Scheme 1.5 an enan-tioselectivity of only 77 % ee is obtained, while 94 % ee is reported using K3[Fe(CN)6]as the reoxidant. The lower enantioselectivity can be explained by some involvementof the so-called second catalytic cycle with the intermediate OsVI glycolate being oxi-dized to an OsVIII species prior to hydrolysis (Scheme 1.6) [24].
Nevertheless, the enantioselectivity was improved by applying a higher ligand con-
centration. In the presence of 5 mol% (DHQD)2PHAL a good enantioselectivity of91% ee is observed for a-methylstyrene. Using tert-butylmethylether as the organicco-solvent instead of tert-butanol, 99 % yield and 89 % ee with only 1 mol%(DHQD)2PHAL are reported for the same substrate. This increase in enantioselectiv-ity can be explained by an increase in the concentration of the chiral ligand in the or-ganic phase. Increasing the polarity of the water phase by using a 10 % aqueousNaCl solution showed a similar positive effect. Table 1.1 shows the results of theasymmetric dihydroxylation of various olefins with NaOCl as the terminal oxidant.
Despite the slow hydrolysis of the corresponding sterically hindered OsVI glyco-
late, trans-5-decene reacted fast without any problems. This result is especially inter-esting since it is necessary to add stoichiometric amounts of hydrolysis aids to the di-hydroxylation of most internal olefins in the presence of other oxidants.
With this protocol a very fast, easy to perform, and cheap procedure for the asym-
metric dihydroxylation is presented. 1 Recent Developments in the Osmium-catalyzed Dihydroxylation of OlefinsScheme 1.6 The two catalytic cycles in the asymmetric dihydroxylation
Asymmetric dihydroxylation of different olefins using NaOCl as terminal oxidant a Entry Olefin Time Yield Selectivity ee ee (%) (h) (%) (%) (%) Ref. a Reaction conditions: 2 mmol olefin, 0.4 mol% K2[OsO2(OH)4], 5 mol% (DHQD)2PHAL, 10 mL H2O,
10 mL tBuOH, 1.5 equiv. NaOCl, 2 equiv. K2CO3, 0 8C. 1.2 Environmentally Friendly Terminal OxidantsEntry Olefin Time Yield Selectivity ee ee (%) (h) (%) (%) (%) Ref. b 5 mol% (DHQD)2PYR instead of (DHQD)2PHAL.
1.2.3 Oxygen or Air
In the past it has been demonstrated by several groups that in the presence of OsO4and oxygen mainly non-selective oxidation reactions take place [25]. However, in1999 Krief et al. published a reaction system consisting of oxygen, catalytic amountsof OsO4 and selenides for the asymmetric dihydroxylation of a-methylstyrene underirradiation with visible light in the presence of a sensitizer (Scheme 1.7) [26]. Here,the selenides are oxidized to their oxides by singlet oxygen and the selene oxides areable to re-oxidize osmium(VI) to osmium(VIII). The reaction works with similaryields and ee values to those of the Sharpless-AD. Potassium carbonate is also used,but only one tenth of the amount present in the AD-mix. Air can be used instead ofpure oxygen. Scheme 1.7 Osmium-catalyzed dihydroxylation using 1O2 and benzyl phenyl selenide
The reaction was extended to a wide range of aromatic and aliphatic olefins [27]. It
was shown that both yield and enantioselectivity are influenced by the pH of the re-action medium. The procedure was also applied to practical syntheses of natural pro-duct derivatives [28]. This version of the AD reaction not only uses a more ecologicalco-oxidant, it also requires much less matter: 87 mg of matter (catalyst, ligand, base,
1 Recent Developments in the Osmium-catalyzed Dihydroxylation of Olefins
reoxidant) are required to oxidize 1 mmol of the same olefin instead of 1400 mgwhen the AD-mix is used.
Also in 1999 there was the first publication on the use of molecular oxygen with-
out any additive to reoxidize osmium(VI) to osmium(VIII). We reported that the os-mium-catalyzed dihydroxylation of aliphatic and aromatic olefins proceeds efficientlyin the presence of dioxygen under ambient conditions [29]. As shown in Table 1.2the new dihydroxylation procedure constitutes a significant advancement comparedwith other reoxidation procedures. Here, the dihydroxylation of a-methylstyrene iscompared using different stoichiometric oxidants. The yield of the 1,2-diol remainsgood to very good (87–96 %), independent of the oxidant used. The best enantioselec-tivities (94–96 % ee) are obtained with hydroquinidine 1,4-phthalazinediyl diether[(DHQD)2PHAL] as the ligand at 0–12 8C (Table 1.2, entries 1 and 3).
The dihydroxylation process with oxygen is clearly the most ecologically favorable
procedure (Table 1.2, entry 5), when the production of waste from a stoichiometricreoxidant is considered. With the use of K3[Fe(CN)6] as oxidant approximately 8.1 kgof iron salts per kg of product are formed. However, in the case of the Krief (Ta-ble 1.2, entry 3) and Bäckvall procedures (Table 1.2, entry 4) as well as in the pre-sence of NaOCl (Table 1.2, entry 6) some byproducts also arise due to the use of co-catalysts and co-oxidants. It should be noted that only salts and byproducts formed
Comparison of the dihydroxylation of a-methylstyrene in the presence of different oxidants
Entry Oxidant Yield Reaction conditions ee TON Waste (oxidant) Ref. (%) (%) (kg/kg diol) a Ligand: Hydroquinidine 1,4-phthalazinediyl diether. b Hydroquinidine p-chlorobenzoate. c K4[Fe(CN)6]. d N-Methylmorpholine (NMM). e PhSe(O)CH2Ph. f NMO/flavin-OOH. g NaCl. 1.2 Environmentally Friendly Terminal Oxidants
from the oxidant have been included in the calculation. Other waste products havenot been considered. Nevertheless the calculations presented in Table 1.2 give arough estimation of the environmental impact of the reaction.
Since the use of pure molecular oxygen on a larger scale might lead to safety pro-
blems it is even more advantageous to use air as the oxidizing agent. Hence, all cur-rent bulk oxidation processes, e. g., the oxidation of BTX (benzene, toluene, xylene)aromatics or alkanes to give carboxylic acids, and the conversion of ethylene intoethylene oxide, use air and not pure oxygen as the oxidant [30]. In Table 1.3 the re-sults of the dihydroxylation of a-methylstyrene as a model compound using air asthe stoichiometric oxidant are shown in contrast to that with pure oxygen (Scheme1.8; Table 1.3) [31]. Scheme 1.8 Osmium-catalyzed dihydroxylation of a-methylstyrene
The dihydroxylation of a-methylstyrene in the presence of 1 bar of pure oxygen pro-
ceeds smoothly (Table 1.3, entries 1–2), with the best results being obtained atpH 10.4. In the presence of 0.5 mol% K2[OsO2(OH)4]/1.5 mol% DABCO or 1.5 mol%(DHQD)2PHAL at pH 10.4 and 50 8C total conversion was achieved after 16 h or 20 hdepending on the ligand. While the total yield and selectivity of the reaction are excel-lent (97 % and 96 %, respectively), the total turnover frequency of the catalyst is com-paratively low (TOF = 10–12 h–1). In the presence of the chiral cinchona ligand
Dihydroxylation of a-methylstyrene with air a Entry Pressure Cat. Ligand L/Os [L] Time Yield Selectivity ee (bar)c (mol%) (mmol L–1) (h) (%) (%) (%)
(DHQD)2PHALe a Reaction conditions: K2[OsO2(OH)4], 50 8C, 2 mmol olefin, 25 mL buffer solution (pH 10.4), 10 mL tBuOH. b 10 mmol olefin, 50 mL buffer solution (pH 10.4), 20 mL tBuOH. c The autoclave was purged with air and then pressurized to the d 1,4-Diazabicyclo[2.2.2.]octane. e Hydroquinidine 1,4-phthalazinediyl diether. 1 Recent Developments in the Osmium-catalyzed Dihydroxylation of Olefins
(DHQD)2PHAL an ee of 80 % is observed. Sharpless et al. reported an enantioselec-tivity of 94 % for the dihydroxylation of a-methylstyrene with (DHQD)2PHAL as theligand using K3[Fe(CN)6] as the reoxidant at 0 8C [32]. Studies of the ceiling ee at 50 8C(88 % ee) show that the main difference in the enantioselectivity stems from thehigher reaction temperature. Using air instead of pure oxygen gas gave only 24 % ofthe corresponding diol after 24 h (TOF = 1 h–1; Table 1.3, entry 3). Although the reac-tion is slow, it is important to note that the catalyst stays active, as shown by the factthat 58 % of the product is obtained after 68 h (Table 1.3, entry 4). Interestingly thechemoselectivity of the dihydroxylation does not significantly decrease after a pro-longed reaction time. At 5–20 bar air pressure the turnover frequency of the catalystis improved (Table 1.3, entries 5–11).
Full conversion of a a-methylstyrene is achieved at an air pressure of 20 bar in the
presence of 0.1 mol% of osmium, which corresponds to a turnover frequency of40 h–1 (Table 1.3, entries 8–11). Thus, by increasing the air pressure to 20 bar, itwas possible to reduce the amount of osmium catalyst by a factor of 5. A decrease ofthe osmium catalyst and the ligand leads to a decrease in the enantioselectivity of from82 % to 62 % ee. This is easily explained by the fact that the ligand concentration deter-mines the stereoselectivity of the dihydroxylation reaction (Table 1.3, entries 7 and 9).
While the reaction at higher substrate concentration (10 mmol instead of 2 mmol)
proceeds only sluggishly at 1 bar even with pure oxygen, full conversion is achievedafter 24 h at 20 bar of air (Table 1.3, entries 10 and 11, and Table 1.4, entries 17 and18). In all experiments performed under air pressure the chemoselectivity of the di-hydroxylation remained excellent (92–96 %).
Table 1.4 shows the results of the osmium-catalyzed dihydroxylation of various ole-
As depicted in Table 1.4 all olefins gave the corresponding diols in moderate to
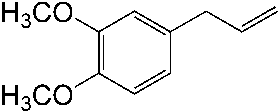
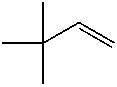
good yields (48–89 %). Applying standard reaction conditions, the best yields of diolswere obtained with 1-octene (97 %), 1-phenyl-1-cyclohexene (88 %), trans-5-decene(85 %), allyl phenyl ether (77 %) and styrene (76 %). The enantioselectivities variedfrom 53 to 98 % ee depending on the substrate. It is important to note that the chemo-selectivity of the reaction decreases under standard conditions in the following sub-strate order: a-methylstyrene = 1-octene > 1-phenyl-1-cyclohexene > trans-5-decene >n-C6F13CH=CH2 > allyl phenyl ether > styrene >> trans-stilbene. A correlation be-tween the chemoselectivity of the reaction and the sensitivity of the produced diol to-wards further oxidation is evident, with the main side reaction being the oxidative clea-vage of the C=C double bond. Aromatic diols with benzylic hydrogen atoms are espe-cially sensitive to this oxidation reaction. Thus, the dihydroxylation of trans-stilbenegave no hydrobenzoin in the biphasic mixture water/tert-butanol at pH 10.4, 50 8Cand 20 bar air pressure (Table 1.4, entry 9). Instead of dihydroxylation a highly selec-tive cleavage of stilbene to give benzaldehyde (84–87 % yield) was observed. Interest-ingly, changing the solvent to isobutyl methyl ketone (Table 1.4, entry 12) makes itpossible to obtain hydrobenzoin in high yield (89 %) and enantioselectivity (98 %) atpH 10.4.
The mechanism of the dihydroxylation reaction with oxygen or air is presumed to
be similar to the catalytic cycle presented by Sharpless et al. for the osmium-cata-
1.2 Environmentally Friendly Terminal Oxidants
Dihydroxylation of various olefins with aira Entry Olefin Cat. Ligand L/Os [L] Time Yield Selectivity ee (mol%) (mmol L–1) (h) (%)b
(DHQD)2PYRg a Reaction conditions: K2[OsO2(OH)4], 50 8C, 2 mmol olefin, 20 bar air, pH = 10.4, 25 mL buffer solution, 10 mL tBuOH; entries 9–12: 15 mL buffer solution, 20 mL tBuOH, entries 17–18: 50 mL buffer solution, 20 mL tBuOH. b Values in par- entheses are for benzaldehyde. c 1 mmol olefin. d pH = 12. e Isobutyl methyl ketone instead of tBuOH. f 10 mmol olefin. g Hydroquinidine 2,5-diphenyl-4,6-pyrimidinediyl diether.
lyzed dihydroxylation with K3[Fe(CN)6] as the reoxidant (Scheme 1.9). The additionof the olefin to a ligated OsVIII species proceeds mainly in the organic phase. De-pending on the hydrolytic stability of the resulting OsVI glycolate complex, the ratedetermining step of the reaction is either hydrolysis of the OsVI glycolate or the reoxi-dation of OsVI hydroxy species. There must be a minor involvement of a second cata-lytic cycle, as suggested for the dihydroxylation with NMO. Such a second cyclewould lead to significantly lower enantioselectivities, as the attack of a second olefinmolecule on the OsVIII glycolate would occur in the absence of the chiral ligand. Theobserved enantioselectivities for the dihydroxylation with air are only slightly lowerthan the data previously published by the Sharpless group, despite the higher reac-tion temperature (50 8C vs. 0 8C). Therefore the direct oxidation of the OsVI glycolateto an OsVIII glycolate does not represent a major reaction pathway. 1 Recent Developments in the Osmium-catalyzed Dihydroxylation of OlefinsScheme 1.9 Proposed catalytic cycle for the dihydroxylation of olefins with OsO4 and oxygen as the terminal oxidant 1.3 Supported Osmium Catalyst
Hazardous toxicity and high costs are the chief drawbacks to reactions using os-mium tetroxide. Besides the development of procedures where catalytic amounts ofosmium tetroxide are joined with a stoichiometrically used secondary oxidant con-tinuously regenerating the tetroxide, these disadvantages can be overcome by theuse of stable and nonvolatile adducts of osmium tetroxide with heterogeneous sup-ports [33]. They offer the advantages of easy and safe handling, simple separationfrom the reaction medium, and the possibility to reuse the expensive transition me-tal. Unfortunately, problems with the stability of the polymer support and leachingof the metal generally occur.
In this context Cainelli and coworkers had already reported, in 1989, the prepara-
tion of polymer-supported catalysts: here, OsO4 was immobilized on several aminetype polymers [34]. Such catalysts have structures of the type OsO4 7 L with theN-group of the polymer (= L) being coordinated to the Lewis acidic osmium center. Based upon this concept, a catalytic enantioselective dihydroxylation was establishedby using polymers containing cinchona alkaloid derivatives [35]. However, since theamine ligands coordinate to osmium under equilibrium conditions, recovery of theosmium using polymer supported ligands was difficult. Os-diolate hydrolysis seemsto require detachment from the polymeric ligand, and hence causes leaching.
Herrmann and coworkers reported on the preparation of immobilized OsO4 on
poly(4-vinyl pyridine) and its use in the dihydroxylation of alkenes by means of hy-drogen peroxide [36]. However, the problems of gradual polymer decomposition andosmium leaching were not solved.
A new strategy was published by Kobayashi and coworkers in 1998: they used mi-
croencapsulated osmium tetroxide. Here the metal is immobilized onto a polymeron the basis of physical envelopment by the polymer and on electron interactionsbetween the p-electrons of the benzene rings of the polystyrene based polymer anda vacant orbital of the Lewis acid [37]. Using cyclohexene as a model compound itwas shown that this microencapsulated osmium tetroxide (MC OsO4) can be usedas a catalyst in the dihydroxylation, with NMO as the stoichiometric oxidant(Scheme 1.10). Scheme 1.10 Dihydroxylation of cyclohexene using microencapsulated osmium tetroxide (MC OsO4)
In contrast to other typical OsO4-catalyzed dihydroxylations, where H2O-tBuOH is
used as the solvent system, the best yields were obtained in H2O/acetone/CH3CN. While the reaction was successfully carried out using NMO, moderate yields wereobtained using trimethylamine N-oxide, and much lower yields were observed usinghydrogen peroxide or potassium ferricyanide. The catalyst was recovered quantita-tively by simple filtration and reused several times. The activity of the recovered cata-lyst did not decrease even after the fifth use.
A study of the rate of conversion of the starting material showed that the reaction
proceeds faster using OsO4 than using the microencapsulated catalyst. This is as-cribed to the slower reoxidation of the microencapsulated osmium ester with NMO,compared with simple OsO4.
Subsequently acryronitrile/butadiene/polystyrene polymer was used as a support
based on the same microencapsulation technique and several olefins, includingcyclic and acyclic, terminal, mono-, di-, tri-, and tetrasubstituted, gave the corre-sponding diols in high yields [38]. When (DHQD)2PHAL as a chiral source wasadded to the reaction mixture enantioselectivities up to 95 % ee were obtained. However, this reaction requires slow addition of the olefin. After running a100 mmol experiment, more than 95 % of the ABS-MC OsO4 and the chiral ligandwere recovered.
Recently Kobayashi and coworkers reported on a new type of microencapsulated
osmium tetroxide using phenoxyethoxymethyl-polystyrene as the support [39]. Withthis catalyst, asymmetric dihydroxylation of olefins has been successfully performedusing (DHQD)2PHAL as a chiral ligand and K3[Fe(CN)6] as a cooxidant in H2O/acet-one (Scheme 1.11). 1 Recent Developments in the Osmium-catalyzed Dihydroxylation of OlefinsScheme 1.11 Asymmetric dihydroxylation of olefins using PEM-MC OsO4
In this instance the dihydroxylation does not require slow addition of the olefin,
and the catalyst can be recovered quantitatively by simple filtration and reused with-out loss of activity.
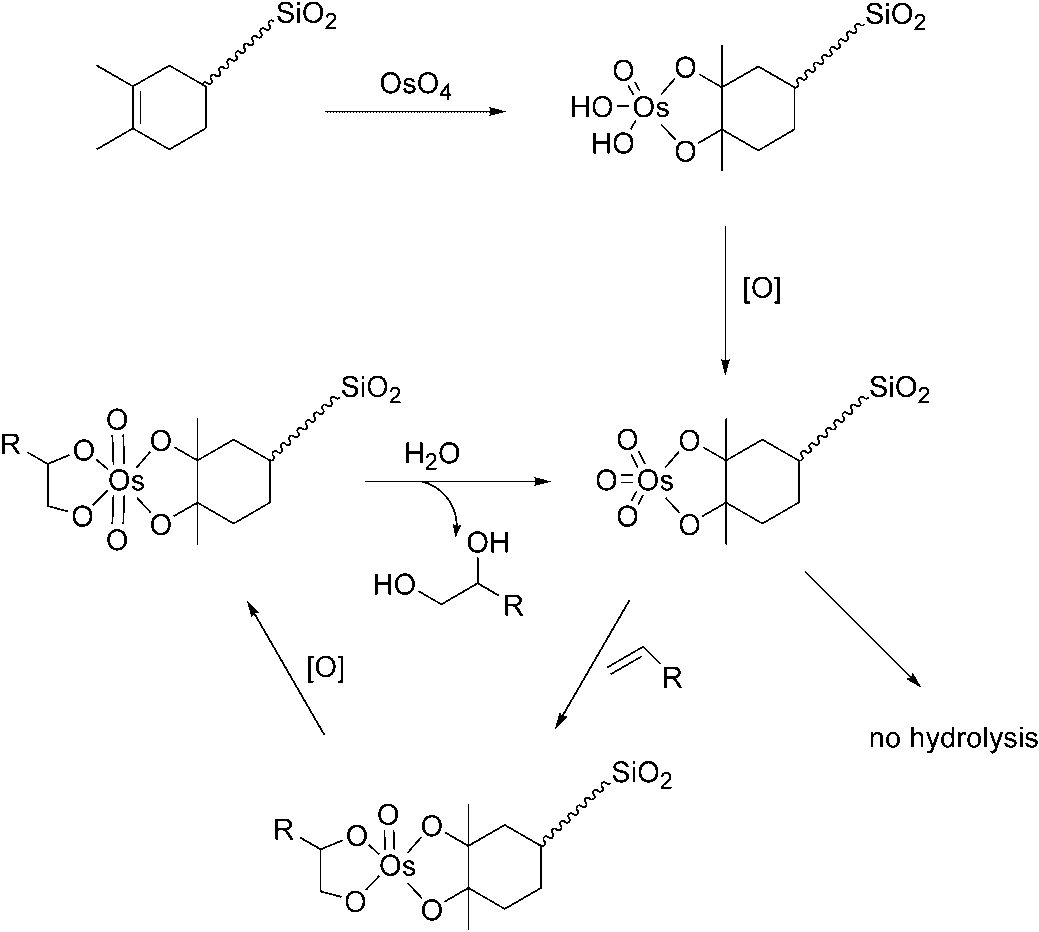
Jacobs and coworkers published a completely different type of heterogeneous os-
mium catalyst. Their approach is based on two details from the mechanism of the cis-dihydroxylation: (1) tetrasubstituted olefins are smoothly osmylated to an osmate(VI)ester, but these esters are not hydrolyzed under mild conditions, and (2) an OsVImonodiolate complex can be reoxidized to cis-dioxo OsVIII without release of the diol;subsequent addition of a second olefin results in an Os bisdiolate complex. Thesetwo properties make it possible to immobilize a catalytically active osmium com-pound by the addition of OsO4 to a tetrasubstituted olefin that is covalently linked toa silica support. The tetrasubstituted diolate ester which is formed at one side of theOs atom is stable, and keeps the catalyst fixed on the support material. The catalyticreaction can take place at the free coordination sites of Os (Scheme 1.12) [40].
The dihydroxylation of monosubstituted and disubstituted aliphatic olefins and
cyclic olefins was successfully performed using this heterogeneous catalyst and
Scheme 1.12 Immobilization of Os in a tertiary diolate complex, and proposed catalytic cycle for cis-dihydroxylation
Rheumatology day unit Dr J Hamilton Open 9am – 5pm Mon – Fri Dr C Heycock Helpline tel: 0191 445 5240 Dr C Kelly Answer phone 24hrs Dr V Saravanan Dr M Rynne Patient information sheet: Minocycline What is Minocycline? Minocycline is an antibiotic that has been used for the treatment of acne, and is also effective in the treatment of rheumatoid ar
1 NAME OF THE MEDICINAL PRODUCT Phenytoin Injection B.P. 250mg/5ml. 2 QUALITATIVE AND QUANTITATIVE COMPOSITION Each 5ml contains Phenytoin Sodium B.P. 250mg. 3 PHARMACEUTICAL FORM Clear, colourless, particle free solution intended for parenteral administration to human beings. 4 CLINICAL PARTICULARS 4.1 Therapeutic indications Phenytoin Injection B.P. is indicated for
 Title: Modern Oxidation Methods
Title: Modern Oxidation Methods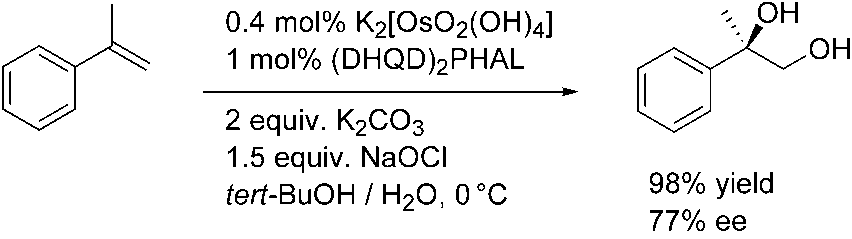 1.2 Environmentally Friendly Terminal Oxidants
1.2.2
1.2 Environmentally Friendly Terminal Oxidants
1.2.2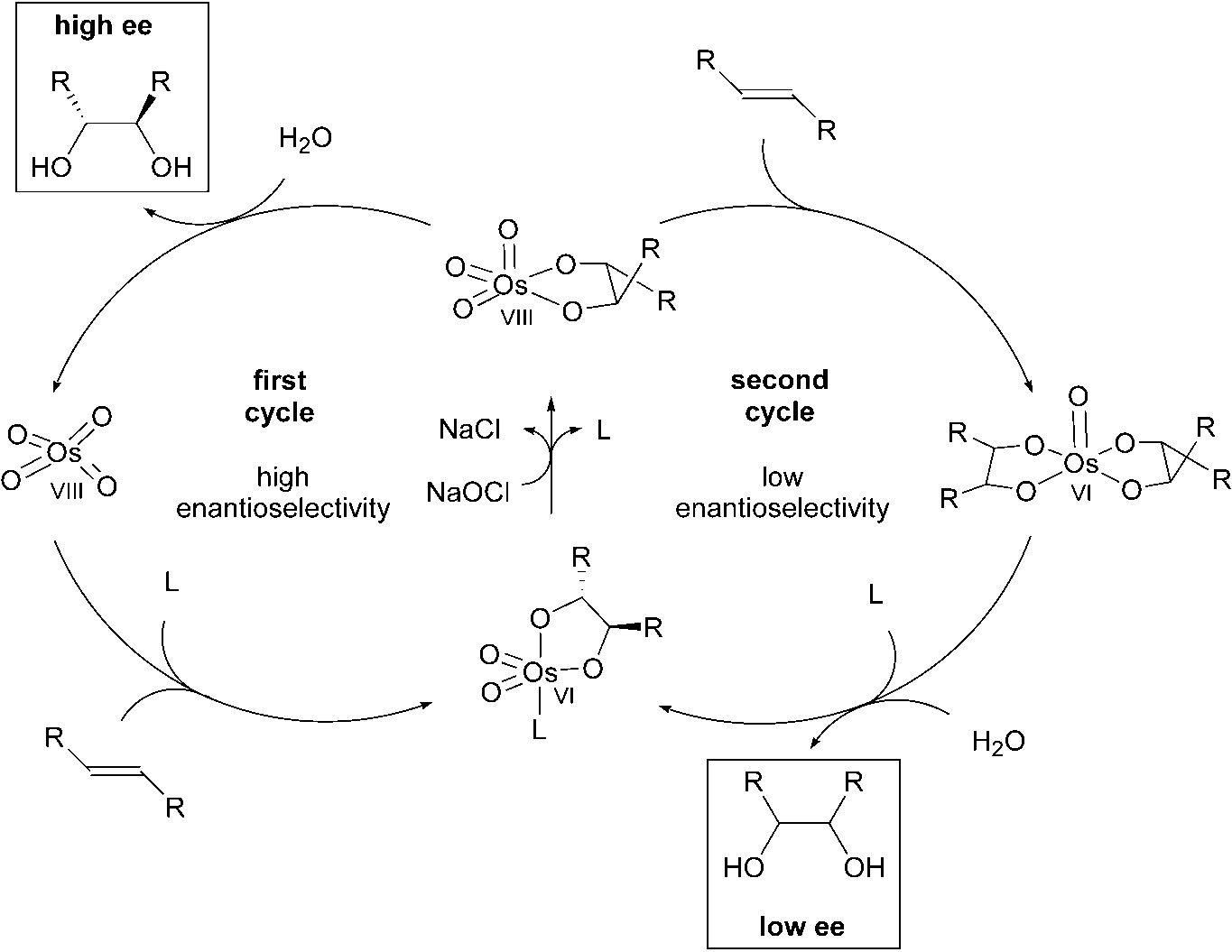

 1 Recent Developments in the Osmium-catalyzed Dihydroxylation of Olefins
Scheme 1.6 The two catalytic cycles in the asymmetric dihydroxylation
1 Recent Developments in the Osmium-catalyzed Dihydroxylation of Olefins
Scheme 1.6 The two catalytic cycles in the asymmetric dihydroxylation
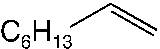
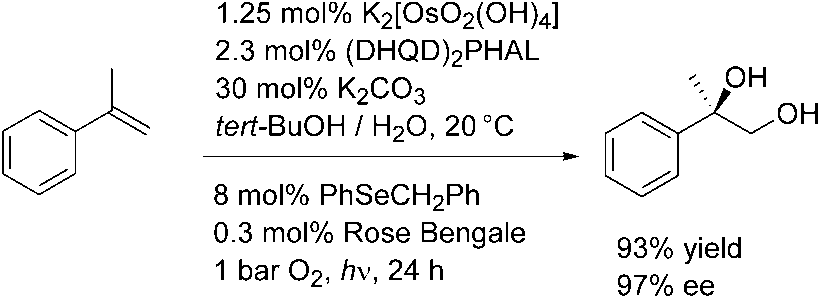 1.2 Environmentally Friendly Terminal Oxidants
Entry
1.2 Environmentally Friendly Terminal Oxidants
Entry 1.2 Environmentally Friendly Terminal Oxidants
from the oxidant have been included in the calculation. Other waste products havenot been considered. Nevertheless the calculations presented in Table 1.2 give arough estimation of the environmental impact of the reaction.
1.2 Environmentally Friendly Terminal Oxidants
from the oxidant have been included in the calculation. Other waste products havenot been considered. Nevertheless the calculations presented in Table 1.2 give arough estimation of the environmental impact of the reaction.
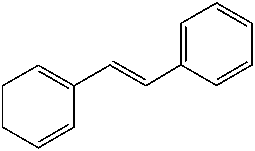

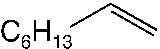
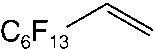 1.2 Environmentally Friendly Terminal Oxidants
Dihydroxylation of various olefins with aira
1.2 Environmentally Friendly Terminal Oxidants
Dihydroxylation of various olefins with aira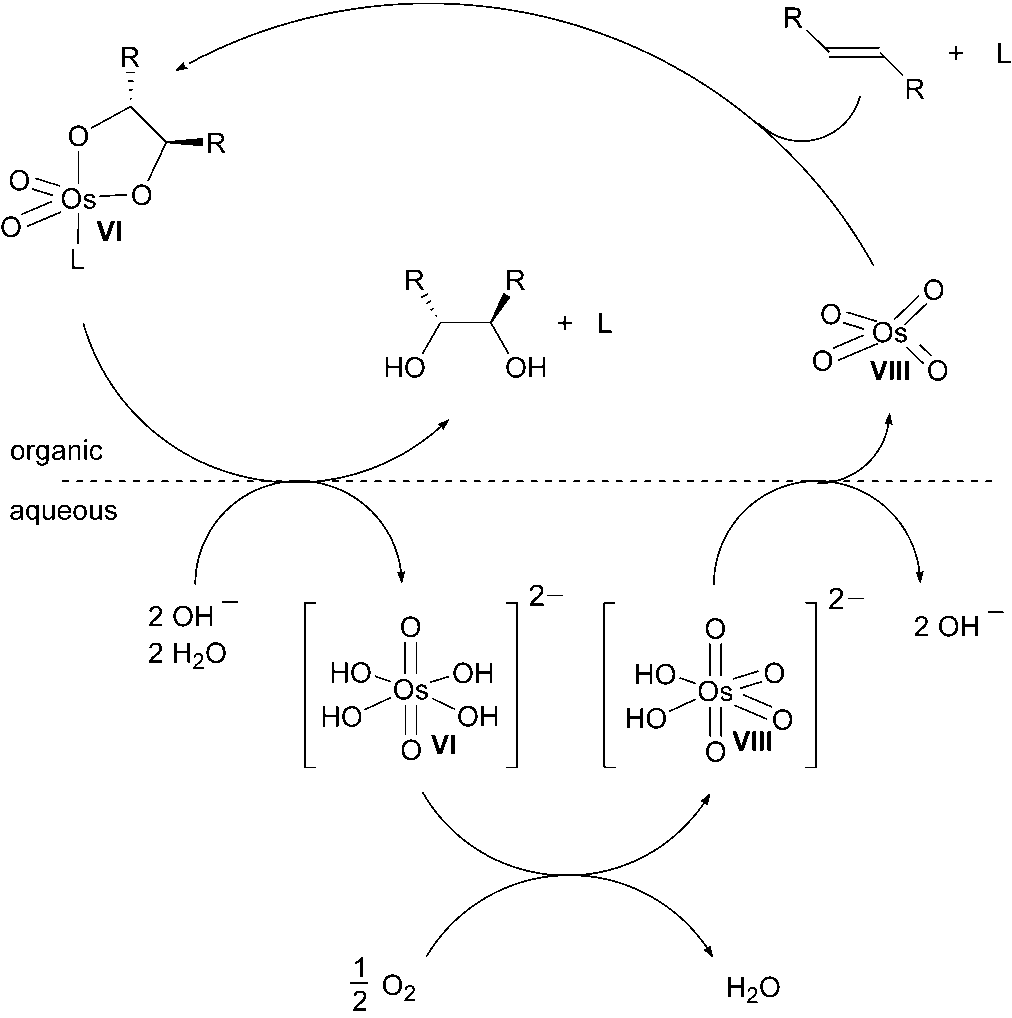 1 Recent Developments in the Osmium-catalyzed Dihydroxylation of Olefins
Scheme 1.9 Proposed catalytic cycle for the dihydroxylation of olefins
1 Recent Developments in the Osmium-catalyzed Dihydroxylation of Olefins
Scheme 1.9 Proposed catalytic cycle for the dihydroxylation of olefins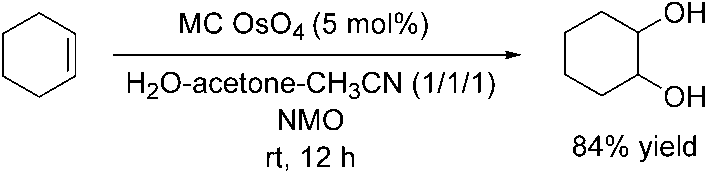 Herrmann and coworkers reported on the preparation of immobilized OsO4 on
poly(4-vinyl pyridine) and its use in the dihydroxylation of alkenes by means of hy-drogen peroxide [36]. However, the problems of gradual polymer decomposition andosmium leaching were not solved.
Herrmann and coworkers reported on the preparation of immobilized OsO4 on
poly(4-vinyl pyridine) and its use in the dihydroxylation of alkenes by means of hy-drogen peroxide [36]. However, the problems of gradual polymer decomposition andosmium leaching were not solved.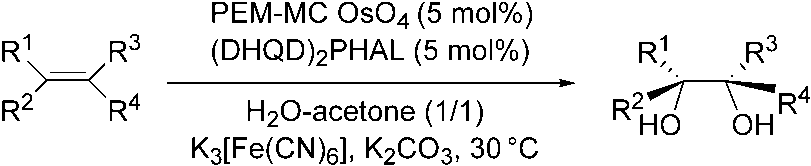
 1 Recent Developments in the Osmium-catalyzed Dihydroxylation of Olefins
Scheme 1.11 Asymmetric dihydroxylation of olefins using
1 Recent Developments in the Osmium-catalyzed Dihydroxylation of Olefins
Scheme 1.11 Asymmetric dihydroxylation of olefins using