REVIEW ARTICLE SEMINARS IN MEDICINE
Mitochondria, which probably evolved from inde-
pendent organisms that became part of the cell, are
able to replicate, transcribe, and translate their DNA
BETH ISRAEL HOSPITAL, BOSTON
independently of nuclear DNA. However, cellular func-tion and mitochondrial function are interdependent.10Nuclear DNA encodes protein subunits of oxidativephosphorylation and the myriad macromolecular com-pounds required for mitochondrial structure and func-tion (e.g., replication, transcription, and translation). Proteins encoded by nuclear DNA must be importedfrom the cytoplasm into the correct position within the
mitochondria.8 During oxidative phosphorylation, ener-
LISA H. UNDERHILL, Assistant Editor
gy is derived from intermediary metabolites to produceATP by means of an electrochemical gradient. This bio-
MITOCHONDRIAL DNA AND DISEASE
chemical process depends on the mitochondrial-DNA–encoded tRNAs that cleave multigene transcripts into
Several features of mitochondrial DNA may be relat-
THE mitochondrial encephalomyopathies are a di- ed to its frequent association with disease. Mitochon-
verse group of disorders that result from the struc-
drial DNA mutates more than 10 times as frequently as
tural, biochemical, or genetic derangement of mito-
nuclear DNA and has no introns, so that a random mu-
chondria.1 Since mitochondrial dysfunction can affect
tation will usually strike a coding DNA sequence. In ad-
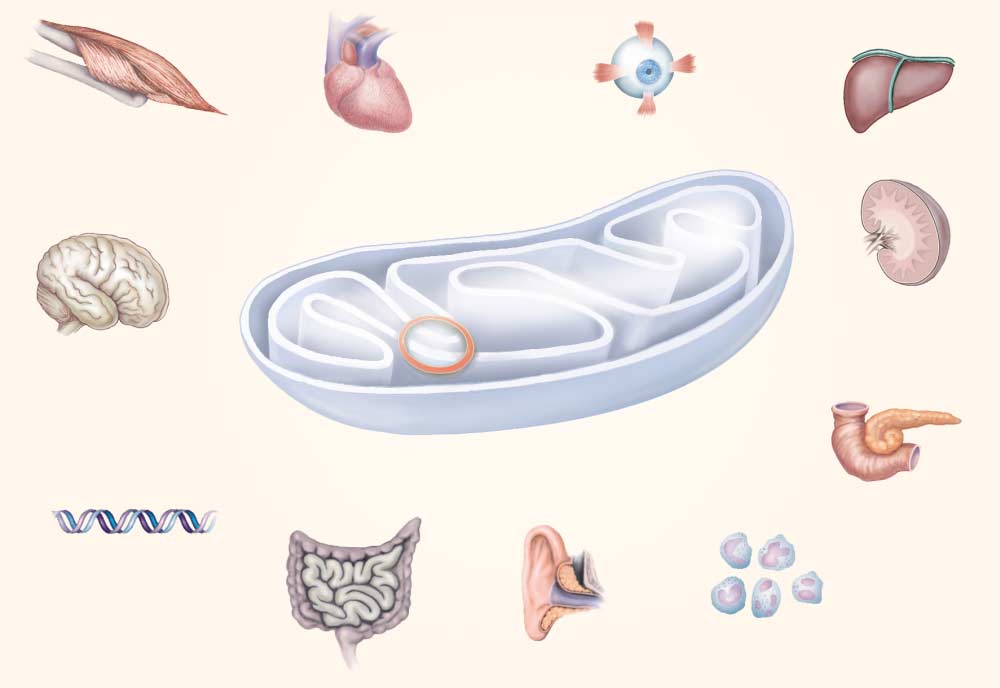
virtually all organ systems (Fig. 1), physicians in many
dition, mitochondrial DNA has neither protective his-
specialties see patients with mitochondrial diseases.
tones nor an effective repair system, and it is exposed
Despite a bewildering array of clinical manifestations
to oxygen free radicals generated by oxidative phospho-
(Tables 1 and 2) and variations in the mode of onset,
course, and progression of disease, many mitochondri-
Mitochondrial DNA is inherited maternally and does
al disorders share prominent systemic effects (Table 2)
not recombine; mutations thus accumulate sequentially
through maternal lineages. Each mitochondrion con-
Our understanding of the role of mitochondrial
tains 2 to 10 DNA molecules, and each cell contains
DNA in certain diseases has evolved rapidly since 1988,
multiple mitochondria. Thus, normal and mutant mi-
when the first mutations in mitochondrial DNA were
tochondrial DNA can coexist within the same cell. This
discovered.2,3 Such mutations have subsequently been
condition, known as heteroplasmy, allows an otherwise
identified in a variety of diseases,4-7 and the pathogenic
lethal mutation to persist. Homoplasmy is the presence
role of cumulative mitochondrial-DNA damage is being
of either completely normal or completely mutant mi-
explored in many common diseases that develop late in
tochondrial DNA. Through the process of replicative
life, and even in the aging process itself.
segregation, the proportions of mutant and normalmolecules can shift as mitochondrial DNA is parti-
STRUCTURE AND FUNCTION OF MITOCHONDRIAL
tioned into daughter cells. Thus, the principles of pop-
ulation genetics, rather than those of mendelian genet-
To understand mitochondrial disease, one must first
ics, govern mitochondrial DNA. Selection pressures
examine the unique features of mitochondrial DNA. Mi-
occur at the molecular and cellular levels, as well as
tochondria generate energy for cellular processes by pro-
at the level of the organism itself. The proportion of
ducing ATP through oxidative phosphorylation. These
mutant mitochondrial DNA required for the occur-
organelles contain their own, extrachromosomal DNA,
rence of a deleterious phenotype, known as the thresh-
which is distinct from DNA in the nucleus.8 Human
old effect, varies among persons, among organ systems,
mitochondrial DNA (known as “the other human ge-
and within a given tissue. The threshold effect depends
nome”) is a double-stranded, circular molecule that en-
on the delicate balance between oxidative supply and
codes 13 protein subunits of 4 biochemical complex-
es and the 24 structural RNAs (2 ribosomal RNAs
The classic mitochondrial phenotypes described be-
[rRNAs] and 22 transfer RNAs [tRNAs]) that are re-
low are caused by gross structural rearrangements (sin-
quired for the intramitochondrial translation of the pro-
gle deletions, multiple deletions, or duplications) or by
tein-coding units.9 Mitochondrial-DNA mutations have
point mutations in mitochondrial DNA. Mutations with
been found in each type of mitochondrial gene (Fig. 2).
the potential to cause a lethal impairment of oxidativephosphorylation (gross structural defects or point mu-
From the Division of Neuromuscular Disease, Department of Neurology, Beth
tations in critical regions) are viable only if they are
Israel Hospital and Harvard Medical School, Boston. Address reprint requests to
heteroplasmic. The majority of the milder, missense
Dr. Johns at Harvard Medical School, Bldg. B1-242, 220 Longwood Ave., Bos-
mutations in protein-coding genes are homoplasmic.
Supported by a grant (R01-EY10864) from the National Eye Institute.
Although mutations have been found in each type of
Downloaded from www.nejm.org on December 19, 2008 . Copyright 1995 Massachusetts Medical Society. All rights reserved.
SEMINARS IN MEDICINE OF THE BETH ISRAEL HOSPITAL, BOSTON
Skeletal muscle Pancreas Nuclear DNA Inner ear
Figure 1. Interaction between Genes Encoded by Nuclear DNA and Those Encoded by Mitochondrial DNA in Oxidative
The intricate function of the oxidative-phosphorylation complexes can be disrupted by defects in the subunits encoded by nuclear DNAand mitochondrial DNA or by defects in intergenomic communication between the two types of DNA. The resulting deficits in the pro-duction of ATP have deleterious effects on a number of organ systems, causing the disorders shown. The red bar indicates the site
of defects in intergenomic communication (depletion and multiple deletions of mitochondrial DNA).
mitochondrial-DNA gene, tRNA mutations predomi-
but they were the first molecularly defined examples of
nate in the phenotypes of mitochondrial encephalomy-
many cardinal neurologic diseases, including stroke
opathy, and protein-coding gene mutations predomi-
(the syndrome of mitochondrial encephalomyopathy,
nate in Leber’s hereditary optic neuropathy. A recent
lactic acidosis, and stroke-like episodes [MELAS]), sei-
report suggests that a point mutation in the 12S rRNA
zures (the MELAS syndrome and myoclonic epilepsy
gene is associated with both spontaneous and antibiot-ic-associated sensorineural deafness11 (Fig. 2). CLASSIC PHENOTYPES OF MITOCHONDRIAL ENCEPHALOMYOPATHY
Before 1988, when the first abnormal mitochondrial
DNA was identified, many diseases were provisionally
classified as mitochondrial disorders because of abnor-
mal morphologic or biochemical features of mitochon-
dria or a pattern of maternal inheritance. The first di-
rect evidence that mitochondrial DNA was involved in
Elevated levels of cerebrospinal fluid protein
a disease came from two observations: the finding of
large deletions in mitochondrial DNA from patients
with mitochondrial myopathies2 and the detection of a
missense mutation in mitochondrial DNA from pa-
tients with Leber’s hereditary optic neuropathy.3 Over
the next several years, the molecular genetic basis of
the classic mitochondrial encephalomyopathies was
elucidated.4-7 These disorders are relatively uncommon,
Downloaded from www.nejm.org on December 19, 2008 . Copyright 1995 Massachusetts Medical Society. All rights reserved.
patterns.18 The pattern of autosomal inheritance sug-
gests the existence of a nuclear-DNA–encoded genethat influences the structure of normal mitochondrial
DNA. Tissue-specific, autosomally transmitted deple-
tion of mitochondrial DNA has also been reported.18
Molecular genetic methods can reliably detect mito-
chondrial-DNA deletions,12-15 but these methods re-
The MELAS Syndrome
In the MELAS syndrome, seizures and stroke-like
events cause subacute brain dysfunction, cerebral struc-
tural changes, and several other clinical and laboratory
abnormalities (Tables 1, 2, and 3). The disease is inher-
ited maternally, but it may not be evident in relatives
who do not have all the overt symptoms. In 80 percent
of cases, there is a point mutation at nucleotide position
Psychiatric disorder (especially depression)
3243 in the tRNALeu(UUR) gene. Other mutations havealso been found in this gene, which appears to be a com-mon target for pathogenetic mutations. The mutation at
with ragged-red fibers), and optic neuropathy (Leber’s
nucleotide position 3243 apparently has multiple pheno-
typic effects, because it is also associated with nondele-tion chronic progressive external ophthalmoplegia, my-
Chronic Progressive External Ophthalmoplegia
opathy, deafness, diabetes, and dystonia.17,20,21
The main features of chronic progressive external
ophthalmoplegia are ptosis, ophthalmoplegia, and limb
Myoclonic Epilepsy with Ragged-Red Fibers
myopathy, but additional clinical features (Tables 1 and
The syndrome of myoclonic epilepsy with ragged-
2) and the laboratory abnormalities characteristic of mi-
red fibers consists of myoclonus, seizures, cerebellar
tochondrial disorders (Table 3) may also be present.
ataxia, and mitochondrial myopathy, as well as neuro-
Skeletal-muscle biopsies in patients with chronic pro-
logic (Table 1) and laboratory (Table 3) abnormalities
gressive external ophthalmoplegia reveal abnormal pro-
that are common in other mitochondrial encephalomy-
liferating mitochondria that cause ragged-red fibers, a
opathies. Maternal relatives may be asymptomatic or
hallmark of the severe biochemical defects in oxidative
have partial clinical syndromes, including lipomas in
phosphorylation in many mitochondrial encephalomy-
a characteristic “horse collar” distribution and cardio-
opathies. The Kearns–Sayre syndrome, a form of chron-
vascular disease.22,23 Pathogenetic mutations have been
ic progressive external ophthalmoplegia that begins be-
demonstrated at nucleotide positions 8344 and 8356 in
fore the age of 20 years, is characterized by atypical
pigmentary retinopathy, as well as elevated levels of cer-ebrospinal fluid protein, ataxia, and heart block.1
Neuropathy, Ataxia, and Retinitis Pigmentosa and
Most patients with chronic progressive external oph-
Maternally Inherited Leigh Disease
thalmoplegia have large, single deletions in mitochon-
The syndrome of neuropathy, ataxia, and retinitis
drial DNA.2,12-15 Almost all these deletions occur spo-
pigmentosa is characterized by proximal-muscle weak-
radically by an unknown mechanism.15 In most cases,
ness, sensory neuropathy, developmental delay, ataxia,
the junction contains directly repeated sequences, in-
seizures, dementia, and retinal pigmentary degenera-
cluding a molecular “hot spot” that accounts for ap-
tion.24 This maternally inherited disorder is associated
proximately 25 percent of all deletions.13-15 A few pa-
with heteroplasmic missense mutations at nucleotide
tients have partially duplicated mitochondrial-DNA
position 8993 in the ATPase 6 gene.24 Large propor-
molecules.16 Many patients without gross structural ab-
tions of the same mutations are also present in patients
normalities of mitochondrial DNA have a point muta-
with maternally inherited Leigh disease.25 Molecular
studies readily detect these point mutations in mito-chondrial DNA extracted from muscle, blood, or urine. Autosomally Transmitted Multiple Deletions in Mitochondrial DNA Leber’s Hereditary Optic Neuropathy
The syndrome of autosomally transmitted multiple
Leber’s hereditary optic neuropathy, the first disease
deletions in mitochondrial DNA has diverse phenotypic
in humans that was linked to heritable point mutations
manifestations, most of which are variants of chronic
in mitochondrial DNA,3 is the exemplar of mitochon-
progressive external ophthalmoplegia.18,19 Multiple de-
drial disease caused by homoplasmic missense muta-
letions differ from single deletions in their inheritance
tions. The main clinical phenotype is painless, sub-
pattern, location within the mitochondrial genome, and
acute, bilateral visual loss, with central scotomas and
molecular structure. Unlike single deletions, which are
abnormal color vision.26 The mean age at the onset is
almost always sporadic, multiple deletions can be trans-
23 years, and three to four times as many males are af-
mitted in autosomal dominant and autosomal recessive
Downloaded from www.nejm.org on December 19, 2008 . Copyright 1995 Massachusetts Medical Society. All rights reserved.
SEMINARS IN MEDICINE OF THE BETH ISRAEL HOSPITAL, BOSTON
Figure 2. Diagram of Human Mitochondrial DNA and the Most Common Associated Pathogenetic Mutations.
Point mutations in structural and protein-coding genes are shown inside the circle, with the clinical phenotype indicated and the nu-cleotide position of the mutation shown in parentheses. The position of the most common single deletion, which is 5 kilobases (kb)long, and the multiple deletions are indicated by the arcs outside the circle. MERRF denotes myoclonic epilepsy with ragged-red fibers;NARP, neuropathy, ataxia, and retinitis pigmentosa; Leigh, maternally inherited Leigh disease; LHON, Leber’s hereditary optic neu-ropathy; and MELAS, the syndrome of mitochondrial encephalomyopathy, lactic acidosis, and stroke-like episodes. O denotes the
origin of heavy-stranded DNA replication, and O the origin of light-stranded DNA replication. CYT-b denotes the apocytochrome-b
subunit; ND-1, ND-2, ND-3, ND-4L, ND-5, and ND-6, NADH dehydrogenase subunits; CO I, CO II, and CO III, cytochrome-c oxidasesubunits; 12S and 16S, ribosomal RNA subunits; and A6 and A8, ATPase subunits. The large open space at the top (which includes
O ) is the noncoding D (displacement) loop.
Leber’s hereditary optic neuropathy, which bears
otide position 13,708 in subunit 5 of the NADH dehy-
little clinical resemblance to the other mitochondrial
drogenase gene increases significantly in association
diseases, was first classified as such a disease because
with four different primary mutations.31 An X-linked
of its pattern of maternal inheritance.26 The visual
nuclear-DNA factor may explain the marked predomi-
loss appears to involve both genetic and epigenetic fac-
nance of symptomatic Leber’s hereditary optic neurop-
tors.28 The mitochondrial-DNA mutations are hetero-
athy in males, but the evidence is conflicting.29 The
geneous, with mutations in at least eight genes that en-
most important epigenetic factors are consumption of
code subunits of three biochemical complexes. Four
mutations, at nucleotide position 11,778, position 3460,
The diversity of the primary mitochondrial-DNA
position 15,257, and position 14,484, may have primary
mutations in Leber’s hereditary optic neuropathy has
pathogenetic importance29 (Fig. 2). Two other mito-
clinical relevance. For example, the probability of visu-
chondrial-DNA mutations were recently found in cyto-
al recovery varies widely, depending on the mutation.27
chrome-c oxidase subunit III in patients with Leber’s
Some variants of the disorder, such as Leber’s heredi-
tary optic neuropathy plus dystonia33 and subacute op-
Several other mitochondrial-DNA mutations may
tic neuropathy and myelopathy,34 are also associated
have secondary roles in Leber’s hereditary optic neurop-
with specific mitochondrial-DNA mutations.
athy.29 The prevalence of a secondary mutation at nucle-
Leber’s hereditary optic neuropathy illustrates the
Downloaded from www.nejm.org on December 19, 2008 . Copyright 1995 Massachusetts Medical Society. All rights reserved.
Table 3. Possible Laboratory Findings in Patients
associated with this mutation, such as the MELAS syn-
drome or nondeletion chronic progressive external oph-thalmoplegia, were not found. Diabetes mellitus, which
Ragged-red fibers in skeletal-muscle–biopsy specimens
is a treatable feature of a number of mitochondrial dis-
Elevated lactate concentrations in serum and cerebrospinal fluid
eases, can thus be the predominant manifestation of a
Axonal and demyelinating peripheral neuropathy on nerve-
Gastrointestinal manifestations of mitochondrial-
Sensorineural hearing loss on audiography
DNA mutations include colonic pseudo-obstruction,19
hepatopathy, and weight loss. The most prominent renal
Basal-ganglia calcification or focal signal abnormalities on
manifestation is a type of nonselective proximal-neph-
Abnormalities on phosphorus-31 nuclear-magnetic-resonance
ron dysfunction, with aminoaciduria, phosphaturia, and
glycosuria, that resembles Fanconi’s syndrome. Lactic
Defective oxidative phosphorylation on biochemical studies
acidosis and an acid–base disturbance or glomerulopa-
Molecular genetic evidence of mitochondrial-DNA mutation
thy may bring patients to the attention of nephrologists. Pearson’s syndrome of exocrine pancreatic dysfunc-tion, a sideroblastic hypoproliferative anemia, and pan-
problems involved in determining the pathogenicity of
cytopenia occurs in association with large-scale, single
any mitochondrial-DNA mutation.35 It can be difficult
deletions in mitochondrial DNA. Other forms of sider-
to link a mutation to a clinical disease for several rea-
oblastic anemia or aplastic anemia may also be associ-
sons: mitochondrial DNA is highly polymorphic, there
ated with acquired or inherited mitochondrial-DNA
is a dissociation between the genotype and the pheno-
mutations. Multiple symmetric lipomas, with their char-
type, different mutations can be associated with the
acteristic horse-collar distribution on the thorax, occur
same phenotype,29 the same mutation can be associat-
in association with the mitochondrial-DNA mutation at
ed with different phenotypes,17 and epigenetic factors
The most prominent pulmonary manifestations of
mitochondrial-DNA mutations are the central hypoven-
SYSTEMIC MANIFESTATIONS OF MITOCHONDRIAL-
tilatory abnormalities of Leigh disease and severe cases
DNA MUTATIONS
of myoclonic epilepsy with ragged-red fibers. Mild ele-
Virtually all tissues in the body depend to some ex-
vations of creatine kinase levels, along with muscle fati-
tent on oxidative metabolism and can therefore be af-
gability, poor stamina, and poor endurance, may bring
fected by mitochondrial-DNA mutations. Patients often
patients with mitochondrial disease to the attention of
first seek care because of the systemic manifestations
rheumatologists, perhaps resulting in an evaluation for
listed in Table 2, which may thus provide the first op-
an inflammatory myopathy. Psychiatric manifestations,
portunity to consider the diagnosis of a mitochondrial
especially depression, have been noted in association
disease. Since these manifestations may be important
with multiple mitochondrial-DNA deletions.37
comorbid features of the disease, they should be diag-nosed and vigorously treated. MITOCHONDRIAL-DNA MUTATIONS IN COMMON
The neurologic manifestations of mitochondrial dis-
DISEASES
orders, involving both the central and the peripheral
One of the next challenges for researchers is to define
nervous systems, have been reported extensively and
the role of mitochondrial-DNA mutations in common
reviewed elsewhere.1,4,6 Less well known are the non-
diseases. The genetic basis of many prevalent diseases is
neurologic manifestations of mitochondrial-DNA mu-
complex and does not follow a simple, single-gene men-
tations. Ophthalmologic manifestations are very com-
delian inheritance. Leber’s hereditary optic neuropathy
mon, involving virtually the entire visual axis from
illustrates the complex interactions between genetic and
the lids, cornea, and extraocular muscles to the occip-
epigenetic factors.28,32 These interactions may involve
ital cortex.36 The cardinal findings include ptosis, oph-
mitochondrial-DNA mutations in subgroups of common
thalmoplegia, optic neuropathy, pigmentary retinopa-
diseases, such as diabetes mellitus, in which the pattern
thy, and cortical visual-field defects.36 Cardiac findings,
of maternal inheritance is not prominent.
which are also common and can be life-threatening, in-
Thus far, I have focused on inherited mutations or
clude cardiomyopathy, conduction disease and heart
gross structural derangements at the gametic level. An-
block, the Wolff–Parkinson–White syndrome, and hy-
other category of mitochondrial-DNA mutations that
may be relevant to late-onset degenerative disorders is
Endocrine manifestations are frequent, and the inci-
the tissue-specific accumulation of somatic (noninherit-
dence of diabetes mellitus is relatively high. Pancreatic
ed) mutations.38 Because of the high rate of mutations
islet cells are extremely active metabolically and are
in mitochondrial DNA, postmitotic tissues or those
thus susceptible to the disruption of oxidative phospho-
with a slow turnover of DNA accumulate the largest
rylation. Diabetes mellitus associated with mitochon-
number of somatic mitochondrial-DNA mutations. Ex-
drial-DNA mutations is mainly due to a defect in insu-
ternal factors may also affect mitochondrial DNA; for
lin secretion. The disorder has been linked with a
example, the antiretroviral drug zidovudine depletes
heteroplasmic point mutation in the tRNALeu(UUR) gene
muscle mitochondrial DNA and causes an acquired mi-
at nucleotide position 3243, usually in association with
sensorineural hearing loss.20 Other neurologic findings
The accumulation of mitochondrial-DNA mutations
Downloaded from www.nejm.org on December 19, 2008 . Copyright 1995 Massachusetts Medical Society. All rights reserved.
SEMINARS IN MEDICINE OF THE BETH ISRAEL HOSPITAL, BOSTON
above a threshold level in certain critical neuronal sub-
DR. JOHNS: I am unaware of any data on this topic,
populations and the consequent deficit in the produc-
but I can speculate that overnutrition may increase ox-
tion of ATP may contribute to the pathogenesis of neu-
idative damage to mitochondrial DNA that results in
rodegenerative diseases,40 particularly those that are
dysfunctional oxidative phosphorylation within mito-
more common with advanced age, such as Alzheimer’s
chondria. These mitochondria may then become the
disease and Parkinson’s disease. Mitochondrial energy
source of excess free radicals that further damage mi-
deficits may contribute to neuronal injury through ex-
tochondrial DNA and other macromolecules in a feed-
Such mechanisms may also contribute to aging itself.
DR. DAVID MOLLER: Because the ratio of mutant
Shigenaga and colleagues have outlined an elegant con-
mitochondrial DNA to the wild type varies so much
cept of aging that incorporates the role of mitochon-
among tissues, how do you go about ruling out a mito-
dria.38 These researchers argue that cumulative, age-
chondrial mutation, especially one that may be inherit-
dependent mitochondrial dysfunction, mediated to a
ed, when you cannot obtain biopsy specimens of the rel-
substantial degree by oxidative damage to mitochon-
evant tissue (e.g., beta cells of the pancreas or the optic
drial DNA and other mitochondrial macromolecules,
nerve)? If you do not have a candidate mutation, how
plays an important part in the aging of cells, tissues,
organ systems, and even the whole organism.
DR. JOHNS: With respect to the availability of DNA
from the relevant tissue, virtually all the point muta-
tions can be detected readily in available tissues, such
Data on the role of mitochondrial-DNA mutations
as leukocytes. Homoplasmic mutations are the same in
in the pathogenesis of several diseases have accumulat-
all tissues. The proportions of heteroplasmic point mu-
ed at a breathtaking pace in the seven years since the
tations may vary according to the type of tissue, but all
first pathogenetic mutations in mitochondrial DNA
that is needed to demonstrate the presence of the mu-
were discovered. Elucidation of the complete mito-
tation is a detectable proportion. Major structural mu-
chondrial-DNA sequence in humans and knowledge
tations, such as deletions, do require the analysis of
about variations in that sequence44 have contributed to
skeletal muscle. There is no easy, overall screening test
a nascent understanding of the clinical implications of
that is sensitive and specific for the presence of mito-
mitochondrial-DNA mutations. The rapid accumula-
chondrial-DNA mutations. Sequencing the entire mito-
tion of disease-specific knowledge about “the other hu-
chondrial-DNA region is impractical, and it is easy to
man genome” may well foreshadow advances in our un-
miss heteroplasmic mutations. My colleagues and I use
derstanding of diseases involving the nuclear genome,
very specific molecular genetic assays for particular
which will emerge from the Human Genome Project.
mutations, and we are interested in developing a meth-
Detailed studies of mitochondrial disease have also
od of analyzing larger mitochondrial-DNA regions.
provided insight into fundamental biologic processes,
DR. JOSEPH MAZJOUB: Do particular tissues have
such as oxidative phosphorylation and aging.
characteristic ratios of mutant mitochondrial DNA to
Mitochondrial-DNA mutations appear to cause an
the wild type, or do the ratios vary? If they vary, what
extensive array of disorders. As the number and types
factors control the variation? Is the half-life of a defec-
of mitochondrial diseases increase, internists and sub-
tive mitochondrion any different from that of a normal
specialists will be in a pivotal position to recognize and
treat these diseases. The advances in our understand-
DR. JOHNS: For heteroplasmic mutations, the ratio
ing of the molecular genetic basis of mitochondrial dis-
of normal-to-mutant mitochondrial DNA varies with-
eases have already had a profound effect on the detec-
in a tissue — for example, from fiber to fiber in skel-
tion and evaluation of mitochondrial diseases. A set of
etal muscle. The initial differences within and be-
sensitive and specific molecular genetic tests has been
tween tissues are determined by mitotic segregation
developed for a number of the mitochondrial encepha-
during embryogenesis. These ratios may change over
lomyopathies.45 The unequivocal diagnosis of a mito-
time, depending on a complex interplay of selection
chondrial disease by such methods is obviously the first
pressures at the molecular, organelle, cellular, tissue,
step in appropriate genetic counseling and treatment.
and organismal levels. Mitochondria are not static
Although the advances in treatment have not paral-
structures within a cell; they undergo dynamic morpho-
leled the advances in diagnosis,46 at least we are now
logic changes. I am unaware of any specific data on a
aware of the biochemical targets. Mitochondrial gene
difference in the half-lives of defective and normal mi-
therapy for these disorders must overcome obstacles
that are different from those encountered in the search
DR. FLIER: What is known about the rate of muta-
for effective nuclear gene therapy, but such therapy is
not beyond the realm of possibility.
DR. JOHNS: I am not aware of studies of mitochon-
drial-DNA mutations in normal brown fat. However,
DISCUSSION
the lipomas that occur in association with the mito-
DR. JEFFREY FLIER: Undernutrition is known to pro-
chondrial-DNA mutation at position 8344 are found in
tect against aging and nuclear-DNA mutations. Does
the areas where brown fat is more abundant.
overnutrition damage mitochondrial DNA, and if so,
DR. CHAIM MAYMAN: What is the role of mitochon-
could this process be related to any of the pathogenic
drial-DNA mutations in the optic neuropathies associ-
Downloaded from www.nejm.org on December 19, 2008 . Copyright 1995 Massachusetts Medical Society. All rights reserved.
DR. JOHNS: The presence of mitochondrial-DNA
17. Moraes CT, Ciacci F, Silvestri G, et al. Atypical clinical presentations asso-
mutations in patients with tobacco–alcohol amblyopia
ciated with the MELAS mutation at position 3243 of human mitochondrialDNA. Neuromuscul Disord 1993;3:43-50.
illustrates the interplay between genetic and epigenetic
18. Zeviani M. Nucleus-driven mutations of human mitochondrial DNA. J In-
factors.32 The mutations appear to confer a genetic sus-
19. Johns DR, Threlkeld AB, Miller NR, Hurko O. Multiple mitochondrial DNA
ceptibility to the deleterious effects of tobacco, alcohol,
deletions in myo-neuro-gastrointestinal encephalopathy syndrome. Am J
or both on the optic nerve. However, not all patients
with tobacco–alcohol amblyopia have detectable mito-
20. van den Ouweland JMW, Lemkes HHPJ, Ruitenbeek W, et al. Mutation in
mitochondrial tRNA(Leu)(UUR) gene in a large pedigree with maternally trans-
chondrial-DNA mutations, and my colleagues and I
mitted type II diabetes mellitus and deafness. Nat Genet 1992;1:368-71.
postulate that other mutations are present elsewhere in
21. Johns DR, Plotkin GM, Logigian EL, Sudarsky LR. Dystonia as a manifes-
tation of the 3243 mtDNA mutation. Ann Neurol 1994;36:315. abstract.
22. Silvestri G, Ciafaloni E, Santorelli FM, et al. Clinical features associated
DR. LAKSHMI KANTHAM: Are there animal models
with the A→G transition at nucleotide 8344 of mtDNA (“MERRF muta-
23. Austin SG, Thandroyen FT, Hecht JT, Vriesendorp FJ, Jones OW, Johns DR.
DR. JOHNS: No. There is a large gap in our knowl-
Expanding the phenotype of the 8344 tRNAlys mitochondrial DNA mutation.
edge of these diseases. We have extensive knowledge
Neurology 1994;44:Suppl 2:A403. abstract.
at the molecular and in vitro cellular levels (rho0 cell
24. Holt IJ, Harding AE, Petty RKH, Morgan-Hughes JA. A new mitochondrial
disease associated with mitochondrial DNA heteroplasmy. Am J Hum Genet
lines), and we also have extensive knowledge of the
clinical phenotypes in humans. However, we have few
25. Santorelli FM, Shanske S, Macaya A, DeVivo DC, DiMauro S. The mutation
data on organ systems and no data obtained under ex-
at nt 8993 of mitochondrial DNA is a common cause of Leigh’s syndrome. Ann Neurol 1993;34:827-34.
perimental conditions. The rho0 human cell lines, which
26. Nikoskelainen EK, Savontaus M-L, Wanne OP, Katila MJ, Nummelin KU.
are depleted of endogenous mitochondrial DNA and
Leber’s hereditary optic neuroretinopathy, a maternally inherited disease: agenealogic study in four pedigrees. Arch Ophthalmol 1987;105:665-71.
repopulated with mutant mitochondria, have been in-
27. Johns DR. Genotype-specific phenotypes in Leber hereditary optic neurop-
dispensable systems for pathophysiologic studies at the
28. Johns DR, Smith KH, Miller NR, Sulewski ME, Bias WB. Identical twins
who are discordant for Leber’s hereditary optic neuropathy. Arch Ophthal-
DR. COLIN M. COSSI: What do you think is the cur-
rent level of public and medical awareness of the mito-
29. Newman NJ. Leber’s hereditary optic neuropathy: new genetic consider-
30. Johns DR, Neufeld MJ. Cytochrome c oxidase mutations in Leber hereditary
DR. JOHNS: I believe that we are at a crucial juncture
optic neuropathy. Biochem Biophys Res Commun 1993;196:810-5.
in the perception of these disorders. The recent an-
31. Johns DR, Neufeld MJ, Park RD. An ND-6 mitochondrial DNA mutation
associated with Leber hereditary optic neuropathy. Biochem Biophys Res
nouncement by the American cyclist Greg Le Mond
that he is retiring from competitive cycling because of
32. Cullom ME, Heher KL, Miller NR, Savino PJ, Johns DR. Leber’s hereditary
a mitochondrial myopathy48 has brought these “myste-
optic neuropathy masquerading as tobacco-alcohol amblyopia. Arch Oph-thalmol 1993;111:1482-5.
rious” disorders to the attention of the public.
33. Jun AS, Brown MD, Wallace DC. A mitochondrial DNA mutation at nucle-
otide pair 14459 of the NADH dehydrogenase subunit 6 gene associated
REFERENCES
with maternally inherited Leber hereditary optic neuropathy and dystonia. Proc Natl Acad Sci U S A 1994;91:6206-10.
1. DiMauro S, Moraes CT. Mitochondrial encephalomyopathies. Arch Neurol
34. Johns DR, Hurko O, Attardi G, Griffin JW. Molecular basis of a new mito-
chondrial disease: acute optic neuropathy and myelopathy. Ann Neurol
2. Holt IJ, Harding AE, Morgan-Hughes JA. Deletions of muscle mitochondrial
DNA in patients with mitochondrial myopathies. Nature 1988;331:717-9.
35. Howell N. Mitochondrial gene mutations and human diseases: a prolegome-
3. Wallace DC, Singh G, Lott MT, et al. Mitochondrial DNA mutation associ-
ated with Leber’s hereditary optic neuropathy. Science 1988;242:1427-30.
36. Johns DR. Mitochondrial DNA mutations and eye disease. In: Wiggs JL,
4. Wallace DC. Diseases of the mitochondrial DNA. Annu Rev Biochem 1992;
ed. Molecular genetics of ocular disease. New York: Wiley–Liss, 1995:201-
5. Brown MD, Wallace DC. Molecular basis of mitochondrial DNA disease.
37. Suomalainen A, Majander A, Haltia M, et al. Multiple deletions of mito-
chondrial DNA in several tissues of a patient with severe retarded depression
6. Schon EA, Hirano M, DiMauro S. Mitochondrial encephalomyopathies:
and familial progressive external ophthalmoplegia. J Clin Invest 1992;90:61-
clinical and molecular analysis. J Bioenerg Biomembr 1994;26:291-9.
7. Howell N. Primary LHON mutations: trying to separate “fruyt” from “chaf.”
38. Shigenaga MK, Hagen TM, Ames BN. Oxidative damage and mitochondrial
decay in aging. Proc Natl Acad Sci U S A 1994;91:10771-8.
8. Attardi G, Schatz G. Biogenesis of mitochondria. Annu Rev Cell Biol 1988;
39. Dalakas MC, Illa I, Pezeshkpour GH, Laukaitis JP, Cohen B, Griffin JL. Mi-
tochondrial myopathy caused by long-term zidovudine therapy. N Engl J
9. Anderson S, Bankier AT, Barrell BG, et al. Sequence and organization of the
human mitochondrial genome. Nature 1981;290:457-65.
40. Shoffner JM, Brown MD, Torroni A, et al. Mitochondrial DNA variants ob-
10. Clayton DA. Structure and function of the mitochondrial genome. J Inherit
served in Alzheimer disease and Parkinson disease patients. Genomics 1993;
11. Prezant TR, Agapian JV, Bohlman MC, et al. Mitochondrial ribosomal RNA
41. Beal MF. Does impairment of energy metabolism result in excitotoxic neu-
mutation associated with both antibiotic-induced and non-syndromic deaf-
ronal death in neurodegenerative illnesses? Ann Neurol 1992;31:119-30.
42. Beal MF, Hyman BT, Koroshetz W. Do defects in mitochondrial energy me-
12. Johns DR, Hurko O. Preferential amplification and molecular characteriza-
tabolism underlie the pathology of neurodegenerative diseases? Trends
tion of junction sequences of a pathogenetic deletion in human mitochondri-
43. Mecocci P, MacGarvey U, Beal MF. Oxidative damage to mitochondrial
13. Johns DR, Rutledge SL, Stine OC, Hurko O. Directly repeated sequences
DNA is increased in Alzheimer’s disease. Ann Neurol 1994;36:747-51.
associated with pathogenic mitochondrial DNA deletions. Proc Natl Acad
44. Cann RL, Stoneking M, Wilson AC. Mitochondrial DNA and human evolu-
14. Schon EA, Rizzuto R, Moraes CT, Nakase H, Zeviani M, DiMauro S. A di-
45. Johns DR, Neufeld MJ. Pitfalls in the molecular genetic diagnosis of Leber
rect repeat is a hotspot for large-scale deletions of human mitochondrial
hereditary optic neuropathy (LHON). Am J Hum Genet 1993;53:916-20.
46. Johns DR. Mitochondrial encephalomyopathy. In: Johnson RT, Griffin JW,
15. Shoffner JM, Lott MT, Voljavec AS, Soueidan SA, Costigan DA, Wallace
eds. Current therapy in neurologic disease. St. Louis: Mosby–Year Book,
DC. Spontaneous Kearns-Sayre/chronic external ophthalmoplegia plus syn-
drome associated with a mitochondrial DNA deletion: a slip-replication
47. King MP, Attardi G. Human cells lacking mtDNA: repopulation with exog-
model and metabolic therapy. Proc Natl Acad Sci U S A 1989;86:7952-6.
enous mitochondria by complementation. Science 1989;246:500-3.
16. Poulton J. Duplications of mitochondrial DNA: implications for pathogene-
48. Almond E. Le Mond announces retirement. Los Angeles Times. December
sis. J Inherit Metab Dis 1992;15:487-98.
Downloaded from www.nejm.org on December 19, 2008 . Copyright 1995 Massachusetts Medical Society. All rights reserved.
Nieuw Influenza Virus (voorheen Mexicaanse griep) Deze algemene informatiebrief wordt aan u als ouder/verzorger van een kind meegegeven, om geïnformeerd te zijn over het nieuwe influenza virus en de ziekteverschijnselen die hierbij horen. Wat is Nieuwe Influenza A (H1N1)? Nieuwe Influenza A (H1N1) (voorheen Mexicaanse griep of varkensgriep) is griep die veroorzaakt wordt door een
These show how a passage is related to other parts of the Bible. To answer the questions most people ask when reading Scripture the “Quest Study Bible” is by far the best and for comments on the text and parallel scriptures I recommend “The NIV Study Bible”. “In the past, God spoke to our ancestors many times and in many ways / but in these last days He has spoken to us through

 SEMINARS IN MEDICINE OF THE BETH ISRAEL HOSPITAL, BOSTON
Skeletal muscle
SEMINARS IN MEDICINE OF THE BETH ISRAEL HOSPITAL, BOSTON
Skeletal muscle