J. Phys. Chem. B 2004, 108, 17992-18002 Computational Study of γ-Butyrolactone and Li+/γ-butyrolactone in Gas and Liquid Phases Marco Masia* and Rossend Rey Departament de Fı´sica i Enginyeria Nuclear, UniVersitat Polite´cnica de Catalunya,Campus Nord B4-B5, Barcelona 08034, SpainReceiVed: July 8, 2004; In Final Form: September 8, 2004
A comprehensive study of structural and dynamical properties of γ-butyrolactone (GBL) and the extent towhich they are affected in the vicinity of a lithium ion, both in gas and liquid phases, is reported. The isolatedGBL molecule is found to be nonplanar, with a barrier of ≈9 kJ/mol to ring inversion. As expected, thelithium ion coordinates the carbonyl oxygen with an almost collinear configuration relative to the carbon-oxygen bond but with a slight tilting toward the lactone oxygen. This configuration holds for clusters of upto four molecules and in the liquid phase as well (where a tetrahedral first solvation shell is found). A highlevel ab initio vibrational analysis, with a new assignment of bands has been performed, which shows substantialred and blue shifts upon lithium solvation, which decrease in a nontrivial way upon increasing the clustersize. To study the solvent effect of the vibrational spectrum, an accurate intramolecular force field has beendeveloped, based on the concept of relaxed potential energy profiles. The inclusion of stretch and bendanharmonicity is shown to be essential in order to explain, not only the absolute value, but the sign of theshifts, particularly for the carbonyl stretching which is substantially downshifted. The shifts obtained for therest of the bands, together with the diffusion coefficients for bulk GBL and for lithium, are in fair agreementwith experimental results. I. Introduction γ-Butyrolactone (GBL, 4-hydroxybutyric acid gamma-lac-
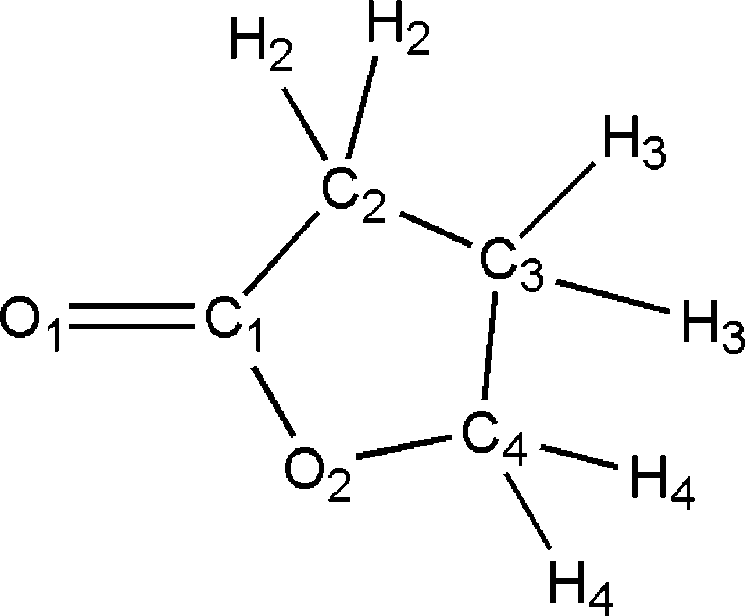
tone, Figure 1), the simplest cyclic ester, is a major chemicalcompound with extensive application in pharmaceuticals,pesticides and petrochemicals.1-6 It is also known to be abuilding block of many natural products of biological activity,like the sesquiterpene lactones, flavor components, alkaloids,antileukemics, and pheromones.8-11 Its biological relevance is
Figure 1. γ-Butyrolactone with the atom labeling used in the text
attributed to its similarity with cyclic peptides.
(notice that hydrogens are grouped in classes).
Recently, GBL has become the focus of increasing techno-
logical interest for its application in lithium ion batteries (LIBs).
Here we have aimed to obtain a comprehensive theoretical
Its physicochemical properties make it suitable to enhance LIBs
understanding at the molecular level: from the isolated molecule
capabilities (reciclability, power, etc.).12 It is an aprotic polar
up to the solvation of the lithum ion in the liquid phase. Both
solvent of moderate viscosity with a dielectric constant of 41.7
ab initio and MD calculations have been used to that purpose.
at ambient temperature, which shows a good solubilizing power
For the gas phase, the optimal structure and vibrational
for lithium salts. Contrary to other good plasticizers employed
frequencies have been computed for the monomer, including a
in LIBs, the liquid-phase exists over a wide range of temper-
complete assignment of bands. Structure and vibrational fre-
atures (the melting and the boiling points are -42 and +206
quencies have also been studied for clusters of Li+, with up to
°C, respectively). Takami et al.6 have recently reported that the
four GBL molecules, as a function of solvation number. Finally,
mixture of GBL with ethylene carbonate (EC) is a promising
and still within the gas phase, an accurate anharmonic intramo-
lecular force field has been developed, following a novel
Despite its importance for basic and applied areas, to our
procedure for parametrization based on the concept of relaxed
knowledge, there are no complete ab initio studies of its structure
potential energy profiles along internal coordinates. Concerning
and vibrational manifold, nor any molecular dynamics (MD)
the liquid state, both neat liquid GBL and Li+ dissolved in GBL
simulation in the liquid phase, particularly in the vicinity of
have been studied. To this end, a standard intermolecular force
the lithium ion. The only theoretical studies to date concern
field has been refined, checking its goodness against counter-
molecular mechanics (MM) calculation of structures,13-15 and
poise corrected potential energy profiles. Finally, a detailed study
ab initio computations of some partial aspects16-20 (see below).
of diffusion and vibrational shifts for molecules within the first
In contrast, and probably due to the aforementioned high
solvation shell of lithium has been performed.
technological impact on LIBs, a substantial amount of experi-
mental work has been reported for Li+-GBL1,2,18,21-28 and for
computational details are described, section III contains the
its mixtures with other plasticizers.6,29
results of the ab initio calculations in the gas phase, and sectionIV contains those for the liquid phase. Finally, the main aspects
* To whom correspondence should be addressed. E-mail: marco.masia@
are summarized in the conclusions section. J. Phys. Chem. B, Vol. 108, No. 46, 2004 17993 TABLE 1: Cartesian Coordinates for the Minimum Energy TABLE 2: Intramolecular Force Field Parameters For Structure, Lennard-Jones Parameters, and Charges for the Stretchingsa Intermolecular Interaction
921.20 -2225.23 3362.30 1.2028 1.211 1.239
Units: [kr ] ) kcal mol-1 Å-i, [r0] ) Å. Comparison of the
equilibrium values with previous studies: molecular mechanics calcula-
tions (MM14), experiment (exp.24), and ab initio (AI18). TABLE 3: Intramolecular Force Field Parameters for Bendingsa II. Computational Details
All ab initio calculations were performed with Gaussian 98.30
Vibrational analysis and geometry optimization were performed
at the MP2 level with the 6-311G basis set augmented with
diffuse and polarization functions.31 The same model chemistry
has been employed for a relaxed potential energy surface scan.
Because of the high memory requirements, the study of the
n]+ with n ranging from 1 to 4 is performed
using the MP2/6-31G model chemistry.
Classical calculations were performed with an in-house MM
code, together with the DL_POLY32,33 suite. The MM code was
used for the scan of the potential energy surface of a single
GBL molecule using a classical intramolecular force field, and
for the vibrational analysis. Finally, the DL_POLY package was
used to perform the liquid-phase simulations. Data analysis
(FFT, curve smoothing, and curve fitting) was performed with
the commercial package Microcal Origin 6.1.34
Units: [kθ ] ) kcal mol-1 rad-i, [θ
equilibrium values with previous studies: molecular mechanics calcula-tions (MM14), experiment (exp.24), and ab Initio (AI18). III. AB Initio Calculations A. Structure 1. 1. Single Molecule. On the experimental side, TABLE 4: Comparison of the Equilibrium Values for the Most Representative Dihedral Angles (degrees) with
infrared,21,24 Raman,24 and microwave spectra22,23,25 of GBL
Previous Studies: Molecular Mechanicsa (MM14) and ab
have been reported. On the other hand, most of the theoretical
Initio (AI18) Calculations
studies correspond to MM calculations (with generic force
fields) of properties such as heats of formation and minimum
energy structures.13-15,17 To our knowledge, previous ab initio
calculations for GBL (using lower levels of theory) were aimed
to study partial aspects such as ring inversion,16 the effect of
isotopic substitution on vibrational circular dichroism,18 intrinsic
basicities,19 and thermal decomposition.20 As a consequence
most of the structural and vibrational measures remain to be
a The sign conventions have been adapted to the ones used here.
In first place, a geometry optimization of the molecule at the
MP2/6-311++G(d,p) level has been performed. The Cartesian
demonstrated that the barrier for inversion of the GBL ring could
coordinates obtained for the minimum energy structure are given
be reliably described using a one-dimensional potential function.
in Table 1. Tables 2-4 contain the equilibrium values obtained
Indeed, a typical double well potential for inversion is obtained
for the internal coordinates, together with those reported in
from a relaxed potential energy scan of the C -
previous works (obtained experimentally,24 with MM methods14
dihedral angle (Figure 2, see details in section III C). Microwave
or with lower level quantum chemical calculations18). A good
spectroscopy measurements25 predict a barrier height for ring
agreement among all results is achieved for bond lengths and
inversion of ≈8.0 kJ mol-1. Our quantum chemical calculation
bending angles, whereas the values for some dihedral angles
produces a slightly higher value (≈9.0 kJ mol-1), with the
show somewhat larger deviations, particularly for the O -
maximum located at 0° (i.e., a planar conformation). This
C3 angle (to our knowledge no experimental results are
conclusion agrees with the expectation of Cremer and Pople in
their study on general monocyclic rings,35 according to which
A basic aspect to consider is that of molecular planarity.
a planar ring should imply a more highly strained ring angle at
Confirming previous works,13,14,16,25 we found that the -carbon
the carbonyl atom than a twisted conformation. Regarding other
lies out of the plane of the remaining four ring atoms resulting
dihedrals (Table 4), our results are very similar to previous ab
in C1 symmetry. With the assumption that the two ring
initio calculations18 but show deviations of up to 8° if compared
puckering coordinates could be treated separately, Lopez et al.25
17994 J. Phys. Chem. B, Vol. 108, No. 46, 2004
only varies by ∼5°. To convey a clearer idea of the changesinduced by the complexation, in Table 5, we report the valuesfor the most affected internal coordinates.
Experimental results obtained with Raman spectroscopy for
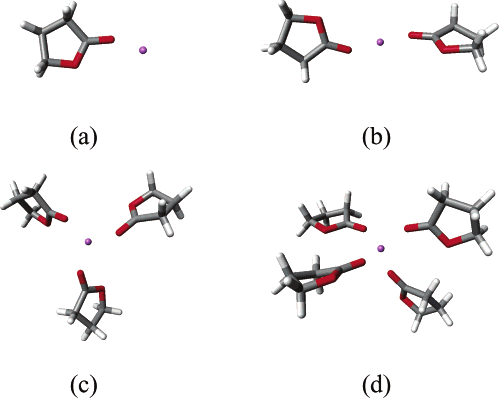
the liquid state suggest that the lithium ion is coordinated byfour GBL molecules37 (a coordination number that has beenfound both for small molecules as water and for larger onessuch as EC). We studied the structure of all GBL complexes(from 1 to 4 molecules plus the lithium ion, Figure 3) with aMP2/6-31G model chemistry (the calculations for the singlemolecule have also been repeated at this lower level of theory,
Figure 2. rPES profile along the C -
to facilitate a consistent comparison along the series). Theminimum energy geometry for the two-coordinated complexhas a linear arrangement with the lithium ion coordinated atopposite sides by the carbonyl oxygens, with the two GBLmolecules lying on perpendicular planes. The three-coordinatedcomplex shows a trigonal configuration with the GBL moleculesslightly tilted to reduce the repulsion. The four-coordinatedcomplex shows a tetrahedral like arrangement as the carbonyloxygens form a dihedral angle of ∼75°. Similar results for thestructure of these complexes where also obtained for EC. As inthat case, distortions of the molecular structure become smallerupon increasing the coordination number, most probably dueto the increasing distance between lithium and the carbonyloxygens. Again, if we compare the distortion induced in thetorsional angle in EC and GBL, we notice that the GBLmolecule is more rigid than EC. Finally, the angle betweencarbonyl axis and the vector joining the ion with the oxygendecreases from ∼157° to ∼140° as the coordination numberincreases, an aspect of interest in the analysis of liquid-phase
Figure 3. γ-Butyrolactone and its complexes [Li(GBL)n]+ with 1 e n e 4. The following colors are assigned to different atomic species:
B. Vibrations. 1. Single Molecule. In Table 6, we report the
red to oxygen, gray to carbon, white to hydrogen, and violet to lithium.
harmonic frequencies obtained from ab initio calculations, those
Some final remarks can be made on the structure: the
obtained with the force field developed in this work (see section
C1) is slightly tilted (3°) with respect
III C), the experimental measures, and, finally, the band
assignment. It is known that the neglect of anharmonicity is a
distance among the two oxygens shorter than the O -
source of disagreement with experimental results, mainly for
separation. For what concerns the hydrogen atoms, differences
high frequency modes. Recently, Scott et Radom38 published
in their distances from the carbons (∼1.09 Å), or in the H-C-H
generic scaling factors for these frequencies so that ab initio
bending angle (∼109°) are negligible.
results can be brought to better agreement with experiment. For
2. [Li(GBL)n]+ (n ) 1-4) Clusters. In a recent study of
MP2/6-311G(d,p) quantum chemical calculations, they proposed
ethylene carbonate,36 a molecule very similar to GBL (the
a scaling factor of 0.9496. Even though our model chemistry is
R-methylene group is substituted by an oxygen), we found that
slightly different (for the inclusion of the ++ diffuse function
the interaction with lithium affects the structure causing the
in the basis set), using the same factor for the highest
distortion of the molecule. A high level calculation (MP2/
frequencies, the corrected ab initio frequencies agree very well
6-31++G(d,p)) of the complex [Li(GBL)]+ has been performed
to look into the most important changes in the equilibriumgeometry of the molecule (Figure 3a). In the previous subsection,
The most recent vibrational analysis is the one by McDer-
it was observed that the carbonyl axis of the single molecule is
mott,24 who used a modified Urey-Bradley force field, with
slightly tilted toward the lactone oxygen; this would suggest
structural assumptions based on experimental measures22,23 and
that the lithium ion might be coordinated by both oxygens if
previous theoretical works.13 Fourteen modes differ from our
the oxygen atoms could get closer upon ion coordination. This
assignment (see Table 6, bold typeface), although only a few
possibility has to be discarded because both the angle among
of them can be considered to be substantial. Particularly
the carbonyl axis and the bisetrix of the C -
stretching, whereas we find that this stretch probably corre-
O2 distance remain fixed. On the other hand, our
calculations clearly show that the lithium ion is only coordinated
sponds to ν17 (what agrees with typical results for lactones39).
1, but still lying out of the carbonyl axis, a muted signal of
2 rocking modes had been assigned to bands for
the presence of the lactone oxygen. Compared to EC, GBL
which we find C-C or O-C stretching modes and vice versa,
seems to be slightly more rigid: coordination affects some bond
a shuffling that can probably be explained if we notice that this
zone of the spectrum is particularly crowded (7 bands in ca.
and dihedral angles are almost unaffected. A representative
300 cm-1). At lower frequencies, we find important differences
example is given by the change of the torsional angle C -
for ν27, ν28, and ν30 which had been previously assigned
O2 upon coordination: when passing from the monomer
respectively to the in-plane ring-CdO torsion, the out of plane,
to the dimer it diminishes by ∼13° in EC, whereas in GBL it
and the in-plane bending of the carbonyl, although here they
J. Phys. Chem. B, Vol. 108, No. 46, 2004 17995 TABLE 5: Values for the Most Affected Coordinates by Ion Coordination Both for High and Low Level Calculations TABLE 6: Vibrational Analysis: High Level Ab Initio, Classical, and Experimental24 Frequencies (cm-1) and Mode Assignmentsa CH2 wagging CH2 twisting C1 O2 stretching CH2 rocking O2 C4 stretching C2 C3 stretching C3 C4 stretching CH2 rocking CH2 rocking ring stretching ring distortion out of plane ring-C1dO1 torsion in plane ring-C1dO1 bending in plane ring-C1dO1 torsion a The results for the mono-coordinated lithium complex are ordered following the assignment for the single molecule. The numbers in brackets
are the high-frequency ab initio scaled values. The shifts with respect to the single molecule are given in the last column (positive sign is used forblueshifts).
are assigned to the out of plane ring-CdO torsion, the in-plane
A preliminary understanding of condensed phase effects might
carbonyl bending and the in-plane ring-CdO torsion, respec-
be obtained from the study of n-coordinated complexes. As it
has been shown in the previous subsection, the structural
changes on the GBL molecule decrease with increasing coor-
n]+ (n ) 1-4) Clusters. As pointed out in
subsection III A the coordination of lithium bears nonnegligible
dination number, an effect that can be expected as well for the
structural changes, what suggests that the strong interaction
vibrational shifts (an issue that was studied in detail for the EC
between GBL and the cation may also induce noticeable shifts
molecule36). According to experimental results,37 the four
of the vibrational frequencies. With high level quantum calcula-
coordinated complex is the most likely in liquid phase. A
tions (MP2/6-311++G(d,p)) substantial shifts (higher than 30
detailed study of the shifts as a function of the coordination
cm-1) have been found for the following modes: ν6, ν7, ν11,
number (with up to four molecules) has been performed with a
ν17, ν25, ν28, and ν30. Actually, these modes are associated with
MP2/6-31G model chemistry. As the number n of coordinating
the most affected degrees of freedom upon ion coordination
molecules increases, also the number m of modes increases
(see section III A). Table 6 contains the shifts for all modes of
(according to m ) 3 × (12 × n + 1) - 6). The majority of
the mono-coordinated complex. It should be noted that a
modes are localized on single molecules so that in a n-
reordering of modes takes place in some cases upon coordina-
coordinated complex one can usually discern n frequencies that
tion. It is the case, for instance, for ν17, which frequency is
can easily be associated to a single mode (the average of these
upshifted by ∼115 cm-1; since this large shift is not experienced
n frequencies is taken as the mode frequency). In some cases,
by ν14-16, it results in a swapping of modes.
there is a nonnegligible dispersion of frequencies (more than
17996 J. Phys. Chem. B, Vol. 108, No. 46, 2004 TABLE 7: Vibrational Analysis: ab Initio Low Level TABLE 8: Intramolecular Force Field Parameters For Frequencies (cm-1) for Single GBL and the Relative Shifts Dihedralsa with its Lithium Complexes [Li(GBL)n]+a
normal mode single GBL ∆ν n ) 1 ∆ν n ) 2 ∆ν n ) 3 ∆ν n ) 4
Units: [An] ) kcal mol-1, [φ0] ) [δ] ) degrees, [kφ] ) kcal mol-1
we add to the view that, given the increased computational
power, force fields tailored to each system can be developed
(at least for molecules of the size of GBL) using as a source of
reference data quantum mechanical results. This is the path
followed for instance to parametrize very flexible force fields
for transition metal complexes, where an accurate description
Positive and negative values of ∆ν correspond to blue and red shifts,
of the quantum mechanical PES far from the minimum is
Recently,36 we applied an efficient methodology to develop
10 cm-1), so that the average value might not be fully
a force field from first principles and applied it to the EC
informative. The carbonyl stretching for the four-coordinated
molecule. The starting point is the usual expansion of the
complex is a relevant example, with frequencies of 1708, 1710,
intramolecular potential in terms of internal coordinates (note
1720, and 1736 cm-1. As it will be shown, this behavior is
that anharmonic terms are included for stretchings and bend-
probably a precursor of the broadening of the absorption band
found in the liquid state, both in experiments and MD simula-tions (see section IV D). Obviously, a subset of modes is
V(r,θ,φ) ) ∑ [k (r - r )2 + k (r - r )3 + k (r - r )4] +
associated to vibrational motion of the whole cluster, and they
have a complex character; most of them fall at wavenumbers
∑ [k (θ - θ )2 + k (θ - θ )3] + ∑ A [1 +
lower than 150 cm-1. An exception corresponds to some
lithium-OdC modes which are found within the range of ring
∑ [k (φ - φ )2] ) V
distortion vibrations; in the four-coordinated complex, there are
cm-1, which will be discussed when the vibrational spectrumfor the liquid phase is addressed.
where r, θ, and φ denote respectively bond lengths, bending
Table 7 illustrates how the shifts become smaller when the
coordination number increases. As will be shown in section IV
The method used to determine the parameters in the previous
D, the results for the four coordinated complex are rather similar
expansion makes use of the relaxed potential energy surface
to those obtained in the liquid phase. Several other features are
(rPES) concept.63 In a rPES scan, the energy is computed along
worth noticing in the shifts experienced by GBL molecules for
a given internal coordinate simultaneously optimizing all the
clusters. One would expect a monotonic variation of the shifts
unconstrained degrees of freedom, so that the minimum total
with the coordination number; remarkably, this is not the case
energy is obtained along the chosen internal coordinate. Such
for many degrees of freedom, as the shifts for the bis coordinated
a procedure can be performed both at the ab initio level and
complex do not follow this trend (see for example ν9, ν14, ν15,
with the classical potential embodied in eq 1. Since the
ν16, ν17, ν23, ν24, ν25, ν26, ν27, ν28, and ν30 in Table 7). Finally,
calculation is done for all internal coordinates, more rPES
the frequency shift decreases at different rates depending on
profiles are obtained than intramolecular degrees of freedom.
the mode, it is not possible to find a simple relation for the
This redundant description indirectly takes into account cross
magnitude of the shift as a function of the coordination number.
effects that are apparently neglected with the functional form
C. Intramolecular Force Field. There are indeed many
used for the potential. The constants in eq 1 are obtained in an
intramolecular force fields available in the literature, like UFF,40
iterative way: after a first guess, the parameter set is refined
AMBER,41-43 MM3,44-51 CHARMM,52,53 OPLS,54-59 and
until the classical rPES profiles reach a good convergence with
COMPASS.60 They can be roughly divided into three classes:
the ab initio ones. Although for the stretching degrees of freedom
(i) generic ones with a large coverage (UFF), (ii) improved
few iterations are required to get a 100% convergence, for
models restricted to some area of applications (e.g., biochem-
bending and torsional coordinates, the fitting procedure is
istry, AMBER, and CHARMM), and (iii) optimized parametri-
slower. The resulting force field is summarized in Tables 2, 3,
zations for condensed matter simulations. In the present work,
and 8. Figure 4 displays some examples of rPES profiles
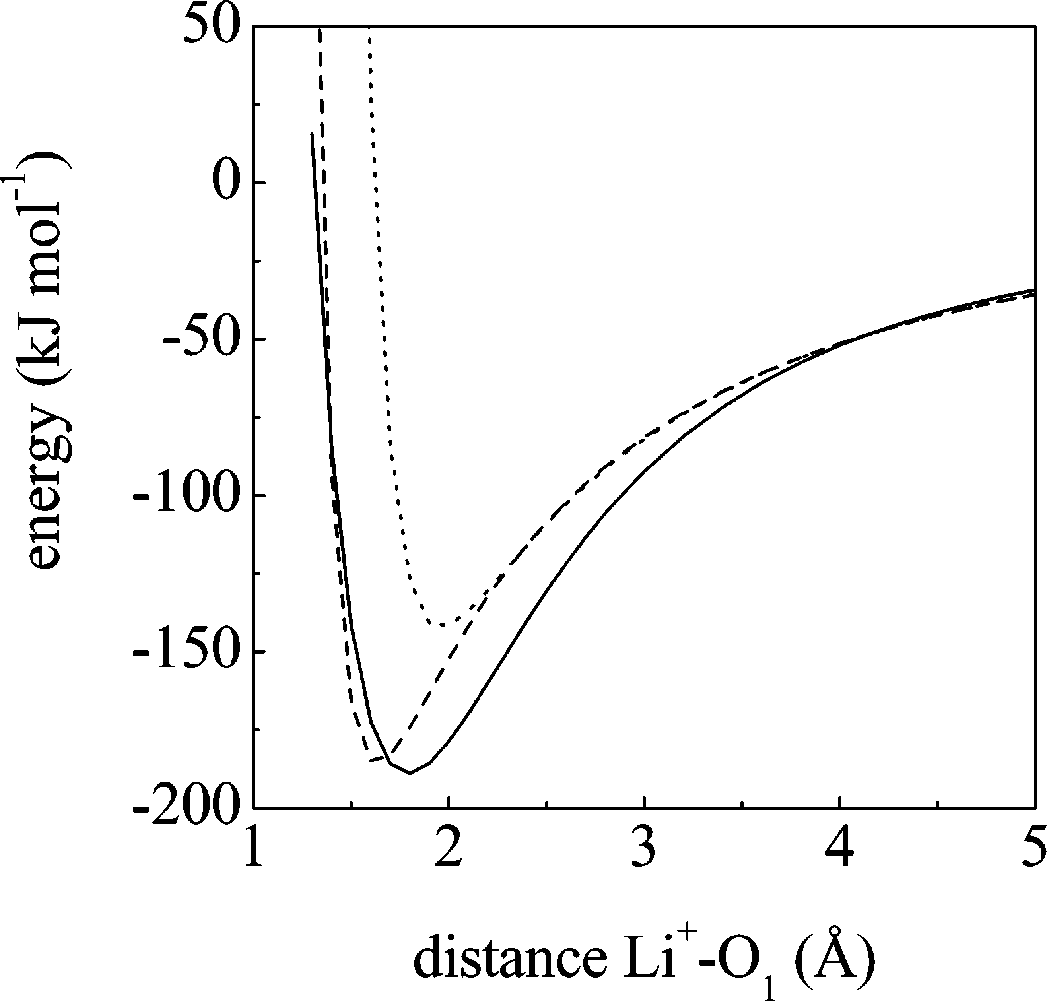
J. Phys. Chem. B, Vol. 108, No. 46, 2004 17997 Figure 5. Potential energy for the GBL-Li+ dimer; ab initio results (solid line), and classical results with the new (dashed line) and old (dotted line) set of Lennard-Jones parameters (see text).
scheme,64,65 constraining them to reproduce the total moleculardipole moment. The latter is slightly overestimated (4.708 Dversus the experimental value of 4.270 D22), which is a desirablefeature in order to balance the absence of polarization effectswith fixed charge models.65 Lennard-Jones parameters for GBLare taken from Carlson et al.66 (with geometric averagecombination rules: σ )
with this parameter set, the diffusion coefficient is lower thanthe experimental one. The origin of this discrepancy lies in theradius taken for H. The value used (σ ) 2.5 Å) is the one typicalfor hydrocarbons, whereas in GBL (and EC) the hydrogens are
Figure 4. rPES profiles along selected internal coordinates: (a) O -
connected to carbon atoms that are near to electron-withdrawing
C1 C2 angle, and (c) C3 C4 O2 C1 dihedral. Filled
groups (carbonate oxygens). This suggests that the electronic
circles, solid line, and dotted line are used respectively for ab initio,our force field, and AMBER results.
cloud for the hydrogen should be smaller. Indeed Sun et al.67have proposed that in the simulation of polycarbonates a valueof σ ) 1.8 Å should be used for hydrogen atoms which are
obtained with ab initio (black circles) and classical calculations
hydrogen bonded to oxygens (it will be shown in the analysis
using both our (solid line) and AMBER (dotted line) force fields.
of liquid structure that the carbonate oxygen tends to bind to
Here AMBER is used as a benchmark since it probably is the
hydrogens). We found that with this smaller hydrogen radius
most popular force field used in atomistic simulations (neverthe-
the diffusion coefficient is very near to the experimental one.
less we have obtained similar results with other force fields such
Along the same line of reasoning it has been found that an
as CHARMM, MM3, and OPLS). Our parametrization produces
optimal value for the lithium ion parameters is σ ) 1.3 Å and
profiles in excellent agreement with the ab initio ones (the same
) 0.191 kcal mol-1. After this parameter fine-tuning, it is
degree of accord is obtained for all intramolecular degrees of
important to check that the modified force field is consistent
freedom, not shown). As mentioned before, our functional form
with ab initio calculations. Figure 5 displays the potential curves
includes the anharmonic terms along stretching and bending
obtained with quantum chemical, and with the modified classical
coordinates. The quantitative importance of an anharmonic
force field just described, for the Li+-GBL dimer. The ab initio
description to better address solvent induced shifts is discussed
result is obtained with a counterpoise68 correction using an MP2/
in section IV D. Panels a and b display the qualitative differences
6-311G(d,p) model chemistry. As can be seen, the refined
in the potential curves when anharmonicity is considered (our
parametrization performs substantially better in reproducing the
force field) and when not (AMBER): the ab initio profile is
interaction between GBL and the lithium ion. Obviously, the
clearly anharmonic. Even for dihedral angles, which are
dimer potential is an approximation to the interaction in the
obviously anharmonic in all force fields, there are noticeable
liquid phase, where many body effects will be present, but we
differences. Panel c shows how AMBER fails to faithfully
do not expect them to be important given the low degree of
association of the neat liquid (see next section).
angle. The vibrational frequencies obtained with the model
All simulations were done in the NVE ensemble with a time
developed here are reported in Table 6, which also contains
step of 0.2 fs. The reference temperature and density were set
the quantum chemical results. The maximum discrepancy with
to 298.15 K and 1.1290 g cm-3 (as reported in the Sigma-
ab initio results is ≈8% (≈15% with AMBER).
Aldrich catalog for the pure product). After an equilibration runof 50 ps, three productions runs of 100 ps each were completed
IV. Molecular Dynamics
to calculate structural and dynamical properties of the system. Two more calculations of 250 ps each were done to compute
A. Simulation Details. Molecular dynamics simulations of
vibrational spectra. For the intramolecular interactions, we used
the pure liquid and of one lithium ion dissolved in liquid GBL
the intramolecular force field developed in section III C and
have been performed. Table 1 contains the parameters used for
the AMBER force field for comparison. The Ewald sum was
the intermolecular potential. Partial charges on the atoms were
employed for electrostatic interactions.
obtained by fitting the electrostatic potential energy surface
B. Structural Properties. 1. Pure GBL. The radial distribu-
(obtained by ab initio MP2/6-311G++(d,p) calculations) at
tion function (RDF) corresponding to the molecular center of
points selected according to the Merz-Singh-Kollman
mass is displayed in panel a of Figure 6. Its overall structure is
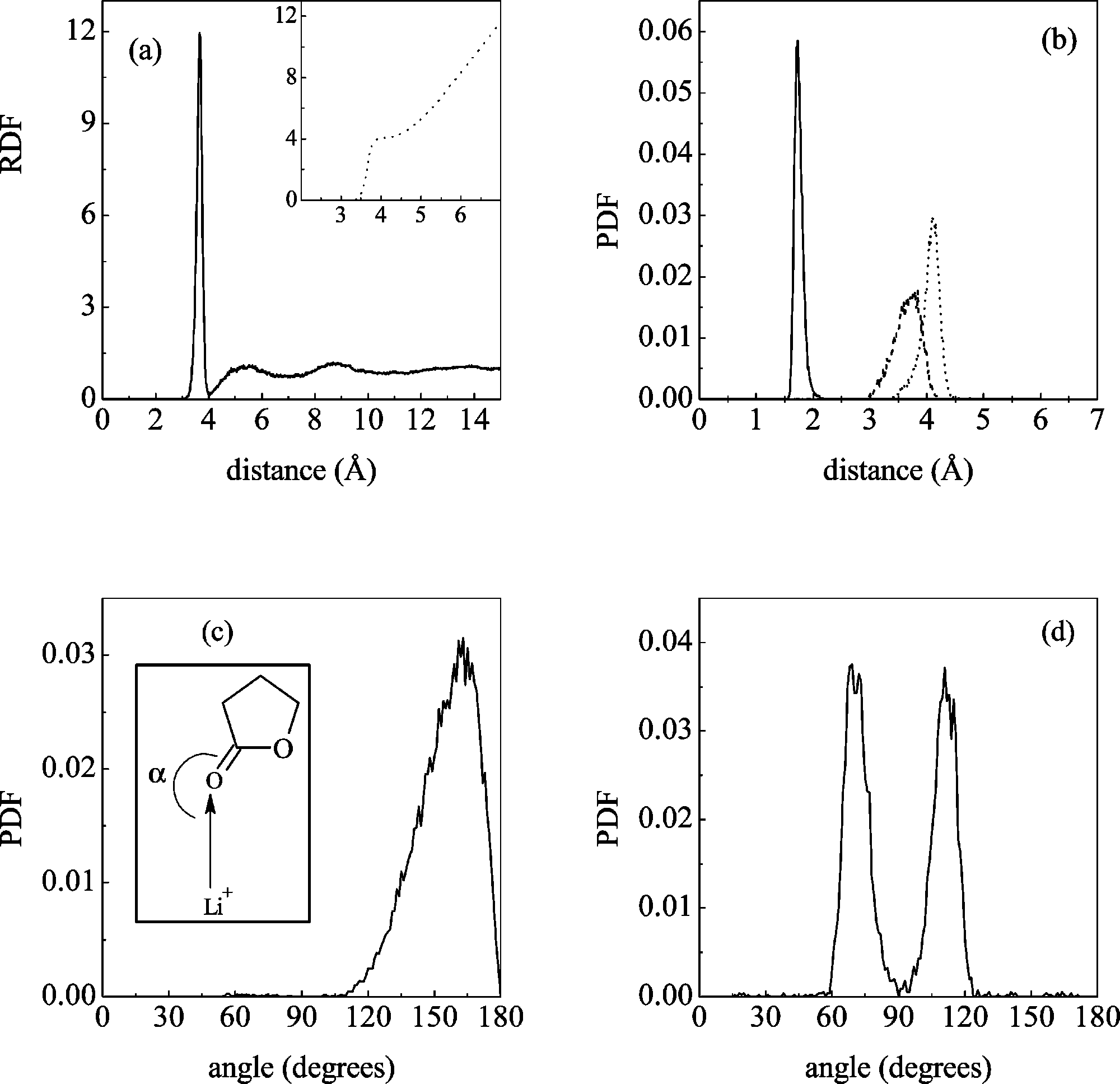
17998 J. Phys. Chem. B, Vol. 108, No. 46, 2004 Figure 7. Structural properties of liquid GBL around lithium ion. (a) Figure 6. Molecular dynamics simulation results: (a) radial distribution
Li+-GBL center of mass radial distribution function, and solvation
function for GBL molecules’ center of mass, (b) and (c) O
number (inset). (b) Probability distribution for the distance Li+-O
(solid line), Li+-O2 (dashed line), and Li+-C2 (dotted line). (c)Probability distribution for R (see inset for definition). (d) Probability
very similar to that of dense simple liquids, what can be further
distribution for the dihedral angle formed by the four carbonyl oxygensnearest to lithium.
confirmed by analysis of the solvation number Ns, defined as
carbonyl oxygen of one molecule would be located midway
N ) 4πF ∫ minr2g(r) dr
between both hydrogens of the C2 group (as a simple geometriccalculation confirms). Such configuration is consistent with the
where g(r) denotes the RDF, F is the number density, and r
lower height of the O1 C2 RDF as compared with those for
is the first minimum of the RDF (7.1 Å). A solvation number
C3 or O1 C4: when coordinating the C2 methylene group
of 12 is found, which is typical of nonassociated liquids.
of one molecule, the carbonyl oxygen of the coordinating
Although this is a signal of a low degree of order, some further
molecule tends to attach preferentially to both hydrogens rather
insight can be obtained from the analysis of partial RDFs.
than directly to the carbon. The peaks located at a shorter
Panel b of Figure 6 displays the O1-oxygen and O1-carbon
distance for the hydrogens belonging to C3 and (to a lesser
radial distribution functions for representative oxygen and
extent) C4 are indicative of a collinear C-H‚‚‚O configuration.
It is also interesting to note the double peak that appears at ≈4
Å, the contact oxygen-oxygen distance, and the same result is
Å in both cases, which is consistent with the distances
O2 (not shown). The corresponding RDFs are
corresponding to the case in which the carbonyl oxygen is
flat and start at larger separations. These features indicate that
coordinated by both C2 hydrogens. In conclusion, this analysis
the oxygens in different molecules tend to stay away from each
points to a substantial amount of hydrogen bonding between
other, what can be explained by the strong electrostatic
the carbonyl oxygen and the methylene hydrogens.
repulsion. Concerning the carbons, the result for O -
2. GBL + Li+. The structural properties of the liquid around
almost identical to those just discussed for the oxygen-oxygen
lithium are collected in Figure 7. The radial distribution function
RDFs, so that the configuration in which the carbonyl oxygen
for the lithium ion is shown in panel a (the inset contains the
would point to C1 of a neighboring molecule is not found. The
solvation number for the first two solvation shells). We find
behavior for the other carbons differs markedly, with a first peak
that the solvation number is exactly four (in accord with the
at the contact oxygen-carbon distance (≈3.2 Å). The slight
experimental estimation37) and that the radius of the first
differences in peak position among different carbons correspond
solvation shell is 4.0 Å. The structure of the complex can be
to their different radius (see Table 1). The results for C4 are
compared to the one obtained with quantum chemical calcula-
not shown for clarity; however, they are very similar to those
tions for clusters (section III A). In panel b, the probability
for C3 but slightly shifted to shorter distances due to the
distribution functions for the distances Li+-O1, Li+-C2, and
somewhat smaller carbon radius. The picture that results is one
Li+-O2 are shown. The most probable distances to O1, O2, and
for which the carbonyl oxygen preferentially solvates the
C2 are respectively 1.73, 3.74, and 4.1 Å: as in the ab initio
methylene groups. This is supported by the analysis of the O -
calculations, the lithium ion is coordinated by the carbonyl
RDFs, with the representative examples displayed in panel c
oxygen and the molecule is tilted allowing the ester oxygen to
of Figure 6. Two rather different behaviors are found: for the
lie nearer to the ion than the R-carbon. To more clearly ascertain
3 and C4, there is a (small) first peak located
the distortion from a linear arrangement of the Li+-O1 C1
at ≈2.4 Å, which corresponds to the contact O-H distance,
atoms, we computed the probability distribution for the angle
(R) formed between the Li+-O1 and the O1 C1 axis (see inset
peak located at a somewhat larger distance (≈2.8 Å). The latter
in panel c for a graphical definition). A maximum exists at
is consistent with a bifurcated configuration in which the
∼160°, well above the result found in the gas phase for the
J. Phys. Chem. B, Vol. 108, No. 46, 2004 17999 Figure 8. Molecular dynamics simulation results for the mean square displacement of GBL molecules’ center of mass (solid line) and of lithium ion (dashed line).
four-coordinated complex (∼140°) and near to the valueobtained for the mono-coordinated one (∼158°). Similar resultswere obtained for the EC case36 and are explained by theattractive interaction with the carbonyl oxygen of second shellmolecules, which tends to draw the methylene groups of firstshell molecules away from the lithium ion, resulting in an angle
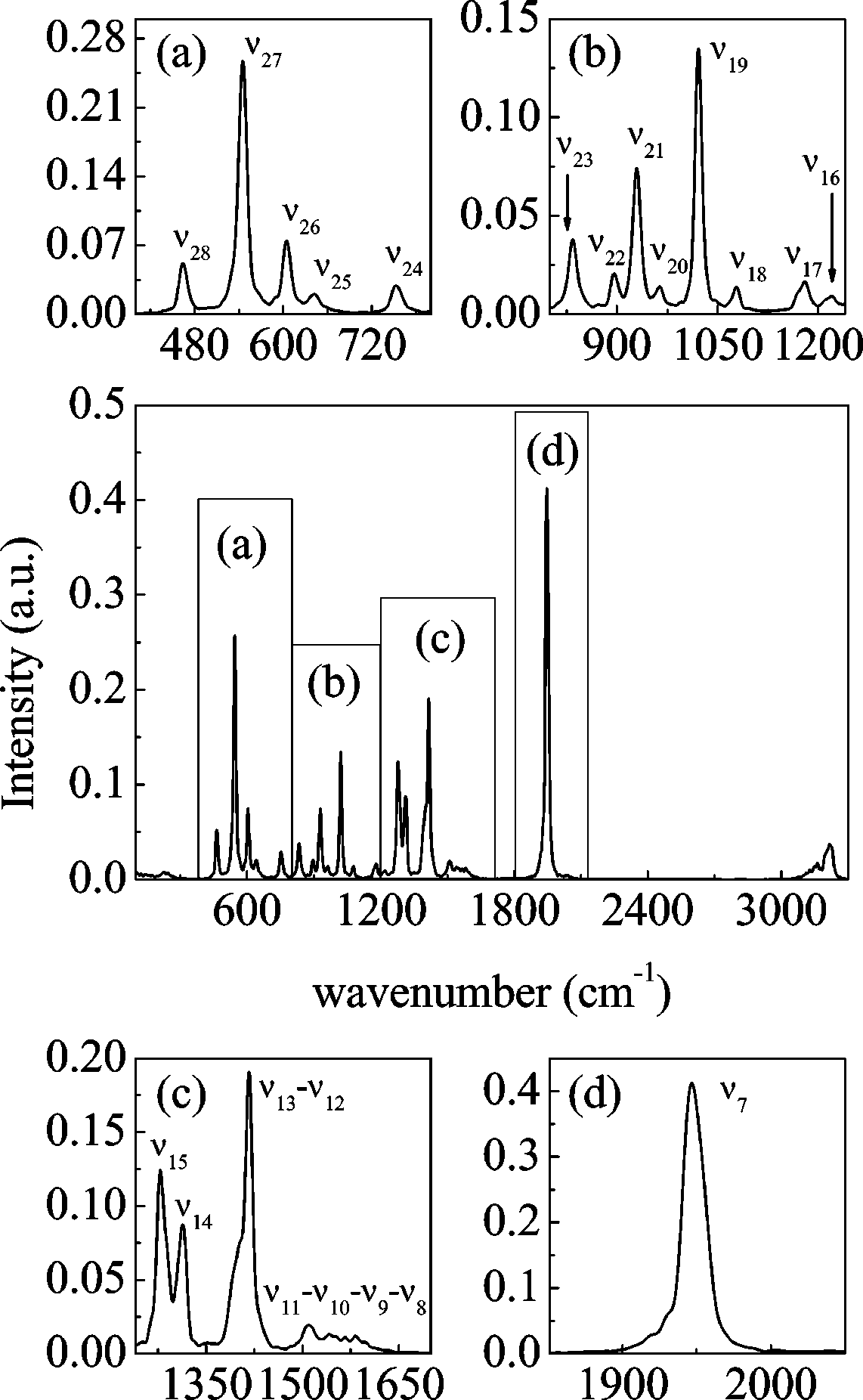
Figure 9. Middle panel: whole vibrational spectrum of pure GBL.
closer to 180°. Finally, the dihedral angle formed by the carbonyl
Smaller panels: details of zones (a), (b), (c), and (d).
oxygens coordinating the cation (last panel) is typical of atetrahedral structure, where the distribution is peaked at ∼71°,
autocorrelation function computed during the simulation (in-
just 4° less than the ab initio result.
cluding all GBL molecules or just those within the first shell
C. Diffusion. Diffusion coefficients are calculated both from
of the ion, see below). According to Berens et al.72-74 the
〈|RB(t) - RB(0)|2〉
S(ω) ) (2π) ∫ dt exp(-iωt)〈M
B denotes the total dipole moment. The shortness of the
and from the velocity autocorrelation function (VACF)
time series available results in a no negligible degree of noise,
so that a filter is required. We used an FFT filter with 20 points
) 1∫ 〈VB(0)‚VB(t)〉 dt
for a correlation function of 100 000 points (we checked in a
previous study36 that this smoothing allows a clearer representa-
where 〈‚‚‚〉 denotes the average for all time origins and all
tion of the spectrum to be obtained without losing important
molecules’ positions (velocities) of the center of mass. The
actual cutoffs used in these formulas are 25 ps (for the MSD,
1. Pure GBL. The whole spectrum of liquid GBL is shown
see Figure 8) and 5 ps (for the VACF integration). The
in the middle panel of Figure 9. Contrary to EC, where a number
experimental value of the GBL diffusion coefficient has been
of bands did not appear in the simulated spectrum,36 here almost
recently measured by means of pulsed gradient spin-echo 1H
all vibrational frequencies are visible. To ease the comparison
NMR,26 and at ambient temperature, it is ≈0.90 × 10-9 m2
with the vibrational analysis done in subsection III B, the
s-1, with which our results agree satisfactorily (D
spectrum is divided into four zones (ν29 and ν30 modes are not
considered because they have a very low intensity). Panel a
s-1). For what concerns lithium diffusion, we found very good
contains all ring modes (ν28 to ν24); of particular intensity is
agreement with experiment as well: Kikuko et al.26 measured
the band for the out of plane ring-CdO torsion (ν27). In panel
a value of ≈0.25 × 10-9 m2 s-1, and we obtain D
b, we show all of the stretching modes for the ring bonds and
the CH2 rocking modes (ν23 to ν16). For what concerns the
s-1. According to Du¨nweg et al.,70 due to the finite size of the
remaining CH2 modes (ν15 to ν8), we can see in panel c that
simulation box, the diffusion coefficient arising from the
the scissoring and the highest frequency wagging modes (ν11
simulation usually underestimates the value for infinite size
to ν8) form a broad band of low intensity where the peaks cannot
systems. They proposed that this could be corrected by adding
be easily distinguished. ν15 to ν12 modes have higher intensity
a constant term ( ) that depends on the simulation box
and two wagging modes coalesce in a single band with a
dimension (L), temperature (T), and viscosity (η)
shoulder due to the ν13 mode. The important carbonyl stretchingmode is depicted in panel d: it shows an asymmetric band which
width at half-height is ∼18 cm-1. Very small shifts (maximum
∼10 cm-1) of the frequencies are noticed if we compare the
condensed phase and the harmonic analysis for the isolated
In our case, considering η ) 1.727 cP,71 we have
molecule. They can be observed mainly in the CH2 twisting
10-9 m2 s-1. Taking into account this correction, the diffusion
and rocking modes and, of minor entity, in ring modes.
coefficients for the pure liquid are even in better agreement with
2. GBL + Li+. To discern the effect of the lithium ion on the
liquid phase spectrum, the dipole moment autocorrelation
D. Vibrational Spectrum. Vibrational spectra were obtained
function was computed during the simulation only for the
by fast Fourier transform (FFT) of the total dipole moment
molecules belonging to the first solvation shell. Figure 10 shows
18000 J. Phys. Chem. B, Vol. 108, No. 46, 2004
blueshifts of respectively ∼27 cm-1 (experimental ∼30 cm-1)and ∼6 cm-1 (experimental ∼10 cm-1). The low intensityobtained for these bands is consistent with experiment as well,as they are only observed at high ionic concentrations. Theremaining modes on which we focus are shown in panel b ofFigure 10. This part of the spectrum seems to be rather sensitiveto coordination. The ν28 mode shows a blueshift of ∼20 cm-1. Experimentally, the presence of a new band whose intensitygrows with salt concentration is observed (with a blueshift of
∼5 cm-1). The ν27 mode is upshifted by ∼15 cm-1, in linewith the experimental blueshift of ∼8 cm-1. In addition, thisband shows a shoulder that might be interpreted as thecontribution of lithium-GBL intermolecular modes: althoughin quantum chemical calculations the majority of intermolecularmodes are found below 150 cm-1, three of them are found inthis zone of the spectrum (see subsection III B), which mightexplain the broad profile of the ν27 mode.
Finally, for what concerns ν24, there is a broadening of the
band whose peak is upshifted by ∼11 cm-1; a blueshift of ∼22cm-1 is found experimentally. Wang et al.27 observed that thecontour becomes more asymmetric as the lithium salt concentra-tion is raised, followed by the splitting of the band at highconcentrations. We should mention that in this zone of the
Figure 10. Comparison between the simulated vibrational spectra of bulk GBL (solid line) and first shell molecules (dashed line) for the
spectrum we also observe the change in intensity of ν26 and
two most representative regions, corresponding respectively to zones
ν25: the former lowers substantially and the latter increases in
(d) and (a) of Figure 9. In panel a, the result with an harmonic force
intensity, while both are slightly upshifted (the entity of these
shifts is within the order of experimental precision). Small shifts(less than 5 cm-1) are also found in all low lying vibrations.
the details of what we called zones a and d for the pure solvent
Even if they are not observed experimentally, this result is
spectrum (previous subsection); both the pure solvent and the
consistent with our ab initio calculation on complexes as
coordinating GBL frequencies are shown. These zones contain
explained in section III B. We can conclude that, as a result of
most of the vibrational frequencies that can be compared with
the strong interaction between lithium and GBL, the most
experimental results: Wang et al.27 found, with IR and Raman
affected vibrational modes are the ring distortions, the methylene
spectroscopy, that the most important shifts correspond to ν7,
rocking and twisting modes, and, obviously, the carbonyl
ν16, ν22, ν24, ν27, and ν28, which will be addressed in turn. It
should be noted that it is not straightforward to compare withexperiment because, in contrast to simulated ones, experimental
V. Conclusions
spectra contain contributions of bulk and ion-coordinatingmolecules. Besides, experimentally there are substantial con-
Concerning structural properties, it has been found in first
tributions from overtone and combination bands (the carbonyl
place that the GBL monomer is nonplanar with a barrier of ≈9
stretching being a prominent example), which in contrast are
kJ/mol for ring inversion, with the carbonyl bond axis slightly
tilted toward the lactone oxygen. This structure is somewhat
The carbonyl stretching normal mode (ν7) is depicted in panel
deformed in the presence of the lithium ion but to a lesser extent
a of Figure 10: upon cation coordination, we notice a redshift
than what is found for instance in the ethylene carbonate case.
of ∼14 cm-1 and a broadening of the spectral band of ∼6 cm-1.
As the number of molecules solvating the ion increases, the
Experimentally, Wang et al. found a bigger broadening and a
distance between the carbonyl oxygen and the ion increases as
shoulder at lower frequencies (which might be indicative of a
well, reducing the molecular distortion. For the important case
shift of ∼24 cm-1). On the other hand, there is a study of Deepa
of the four coordinated cluster, the structure is tetrahedral. In
et al.,29 who found a redshift of 10 cm-1. Our result thus falls
addition, the carbonyl axis is not collinear with the lithium ion,
midway between both experimental estimations. The same panel
but the lactone oxygen is closer to lithium than the R-carbon.
dramatically illustrates the effect of neglecting anharmonicity.
Such a configuration is maintained in the liquid phase, but with
If only the harmonic terms of the force field are considered,
an increased tendency to a collinear configuration due to the
instead of a red shift, a blueshift is obtained. To discard that
attractive effect of second shell molecules. This attraction is
this is not a particular feature of the force field employed, similar
explained by the analysis of radial distribution functions for
simulations have been run using the AMBER force field (which
neat liquid GBL: the carbonyl oxygen tends to solvate the
we recall does not contain any anharmonicity for stretchings or
methylene groups. Particularly, the solvation of the R-carbon
bendings). Again the same result is obtained:
differs from the two other methylene groups in that the oxygen
coordination, the carbonyl stretching mode is upshifted to higher
tends to sit midway between both hydrogens.
wavenumbers. In short, a fully harmonic force field is not able
Given that the main probes of GBL are spectroscopic, a
to reproduce the correct sign of the shift, what should be
special emphasis has been put on vibrational properties, starting
regarded as an important limitation of most force fields if they
with a full new assignment of bands. Substantial shifts have
are to be used to interpret spectroscopic measures of solvated
been found upon lithium coordination. The cases of the C-O
stretches are particularly remarkable for the Li+-GBL dimer:
For what concerns ν16 (CH2 twisting mode) and ν22 (CH2
the carbonyl stretch frequency is downshifted by ≈77 cm-1,
rocking mode), we observed, consistently with experiment,
J. Phys. Chem. B, Vol. 108, No. 46, 2004 18001
linked to the ion) is upshifted by a larger value (≈100 cm-1).
(25) Lopez, J. C.; Alonso, J. L.; Cervellati, R.; Esposti, A. D.; Lister,
Ring modes are substantially affected as well. The shifts
D. G.; Palmieri, P. J. Chem. Soc., Faraday Trans. 1990, 86, 453.
(26) Hayamizu, K.; Aihara, Y.; Arai, S.; Martinez, C. G. J. Phys. Chem.
decrease upon increasing the solvation number but not neces-
B 1999, 103, 519.
sarily in a monotonic way for all modes. This is the case for
(27) Wang, J.; Xuan, X.; Lu, J.; Pei, N.; Mo, Y. Z. Phys. Chem. 2001,
O2 stretch, where the shift is increased for
the trimer compared to the dimer, followed by a gradual decrease
(28) Mandal, S. K.; Amin, A. R.; Crowe, W. E. J. Am. Chem. Soc. 2001,
as the number of GBL molecules is increased. A direct
(29) Deepa, M.; Sharma, N.; Varshney, P.; Agnihotry, S. A.; Chandra,
comparison with experimental results is possible in the liquid
R. Ionics 2000, 6, 408.
phase. To this end, an intramolecular force field has been
(30) Frisch, M. J.; Trucks, G. W.; Schlegel, H. B.; Scuseria, G. E.; Robb,
specially tailored to the GBL molecule, following a procedure
M. A.; Cheeseman, J. R.; Zakrzewski, V. G.; Montgomery, J. A., Jr.;Stratmann, R. E.; Burant, J. C.; Dapprich, S.; Millam, J. M.; Daniels, A.
founded on the concept of relaxed potential energy profiles. This
D.; Kudin, K. N.; Strain, M. C.; Farkas, O.; Tomasi, J.; Barone, V.; Cossi,
new potential includes anharmonic terms for stretches (up to
M.; Cammi, R.; Mennucci, B.; Pomelli, C.; Adamo, C.; Clifford, S.;
quartic contributions) and bends (cubic) and has been shown
Ochterski, J.; Petersson, G. A.; Ayala, P. Y.; Cui, Q.; Morokuma, K.; Malick,D. K.; Rabuck, A. D.; Raghavachari, K.; Foresman, J. B.; Cioslowski, J.;
to be superior to conventional force fields regarding potential
Ortiz, J. V.; Stefanov, B. B.; Liu, G.; Liashenko, A.; Piskorz, P.; Komaromi,
profiles and harmonic frequencies for the monomer. More
I.; Gomperts, R.; Martin, R. L.; Fox, D. J.; Keith, T.; Al-Laham, M. A.;
importantly, the analysis of the carbonyl stretch in the liquid
Peng, C. Y.; Nanayakkara, A.; Gonzalez, C.; Challacombe, M.; Gill, P. M. W.; Johnson, B. G.; Chen, W.; Wong, M. W.; Andres, J. L.; Head-Gordon,
phase has illustrated how the neglect of anharmonic contribu-
M.; Replogle, E. S.; Pople, J. A. Gaussian 98, revision A.11.2; Gaussian,
tions results in a wrong sign for the predicted shift. This is a
critical feature to take into consideration if one wants to use
(31) Frisch, M. J.; Pople, J. A.; Binkley, J. S. J. Chem. Phys. 1984, 80,
generic force fields to theoretically interpret spectroscopic
(32) DL_POLY is a package of molecular simulation routines written
measures. Finally, the calculation of the spectrum for the
by W. Smith and T. R. Forester, copyright The Council For The Central
molecules belonging to the first shell produces results which
Laboratory Of The Research Council, Daresbury Laboratory at Daresbury,
are in fair agreement with experimental shifts. This has allowed
several shoulders and/or broadenings appearing in experimental
(33) http://www.cse.clrc.ac.uk/msi/software/DL_POLY/. (34) http://www.originlab.com.
spectra to be interpreted as being due to lithium induced shifts
(35) Cremer, D.; Pople, J. A. J. Am. Chem. Soc. 1975, 97, 1354.
(36) Masia, M.; Probst, M.; Rey, R. J. Phys. Chem. B 2004, 108, 2016. (37) Caillon-Caravanier, M.; Bosser, G. Claude-Montigny, B.; Lemor-
dant, D. J. Electrochem. Soc. 2002, 149, E340. Acknowledgment. This work was supported by EC TMR
(38) Scott, A. P.; Radom, L. J. Phys. Chem. 1996, 100, 16502.
network HPRN-CT-2000-19 (“Solvation Dynamics and Ionic
(39) Pretsch, E.; Clerk, T.; Seibl, J.; Simon, W. Tablas para la
Mobility in Conventional and Polymer Solvents”) and MCYT
determinacio´n estructural por metodo´s espectroscopicos; Springer-Wer-lag: New York, 1998.
(40) Rappe´, A. K.; Casewit, C. J.; Colwell, K. S.; Goddard, W. A., III;
Skiff, W. M. J. Am. Chem. Soc. 1992, 114, 10024.
(41) Weiner, S. J.; Kollman, P. A.; Nguyen, D. T.; Case, D. A. J.References and Notes Comput. Chem. 1986, 7, 230.
(42) Cornell, W. D.; Cieplak, P.; Bayly, C. I.; Gould, I. R.; Merz, K.
(1) Zhu, Y.-L.; Xiang, H.-W.; Wu, G.-S.; Bai, L.; Li Y.-W. Chem.
M.; Ferguson, D. M.; Spellmeyer, D. C.; Fox, T.; Caldwell, J. W.; Kollman,
Commun. 2002, 3, 254.
P. A. J. Am. Chem. Soc. 1995, 117, 5179.
(2) Tarvainen, M.; Sutinen, R.; Somppi, M.; Paronen, P. Poso A.Pharm. Res. 2001, 18 (12), 1760.
(44) Allinger, N. L.; Yuh, Y. H.; Lii, J.-H. J. Am. Chem. Soc. 1989,
(3) Herrmann, U.; Eming, G. Chem. Eng. Technol. 1998, 21, 285.
(4) Harris, N.; Tuck, M. W. Hydrocarbon Process. 1990, 69, 79.
(45) Lii, J.-H.; Allinger, N. L. J. Am. Chem. Soc. 1989, 111, 8566.
(5) Banker, G. S. J. Pharm. Sci. 1966, 55, 81.
(46) Lii, J.-H.; Allinger, N. L. J. Am. Chem. Soc. 1989, 111, 8576.
(6) Takami, N.; Sekino, M.; Ohsaki, T.; Kanda, M.; Yamamoto, M. J.
(47) Allinger, N. L.; Geise, H. J.; Pyckhout, W.; Paquette, L. A.;
Power Sources 2001, 97-98, 677.
Gallucci, J. C. J. Am. Chem. Soc. 1989, 111, 1106.
(7) Vose, J.; Tighe, T.; Schwartz, M.; Buel, E. J. Forensic Sci. 2001,
(48) Allinger, N. L.; Li, F.; Yan, L. J. Comput. Chem. 1990, 11, 848.
(49) Allinger, N. L.; Li F.; Yan, L.; Tai, J. C. J. Comput. Chem. 1990,
(8) Hoffmann, H. M. R.; Rabe, J. Angew. Chem., Int. Ed. Engl. 1985,
(50) Lii, J.-H.; Allinger, N. L. J. Phys. Org. Chem. 1994, 7, 591.
(9) Koch, S. S. C.; Chamberlin, A. R. J. Org. Chem. 1993, 58, 2725.
(51) Lii, J.-H.; Allinger, N. L. J. Comput. Chem. 1998, 19, 1001.
(10) Brown, H. C.; Kulkarni, S. V.; Racherla, V. S. J. Org. Chem. 1994,
(52) MacKerrell, A. D., Jr.; Bashford, D.; Bellot, M.; Dunbrack, R. L.,
Jr.; Evanseck, J. D.; Field, M. J.; Fisher, S.; Gao, J.; Guo, H.; Ha, S.; Joseph-
(11) Donate, P. M.; Frederico, D.; Da Silva, R.; Constantino, M. G.;
McCarthy, D.; Kuchnir, L.; Kuczera, K.; Lau, F. T. K.; Mattos, C.;
Del Ponte, G.; Bonatto, P. S. Tetrahedron Asym. 2003, 14, 3253.
Michnick, S.; Ngo, T.; Nguyen, D. T.; Prodhom, B.; Reiher, W. E., III;
(12) Chagnes, A.; Carre´ B.; Willmann P.; Dedryve´re R.; Gonbeau D.;
Roux, B.; Schlenkrich, M.; Smith, J. C.; Stote, R.; Straub, J.; Watanabe,
Lemordant, D. J. Electrochem. Soc. 2003, 150 (9), A1255.
M.; Wio´rkiewicz-Kuczera, J.; Yin, D.; Karplus, M. J. Phys. Chem. B 1998,
(13) Allinger, N. L.; Chang, S. H. M. Tetrahedron 1977, 33, 1561.
(14) Allinger, N. L. Pure Appl. Chem. 1982, 54 (12), 2515.
(53) Foloppe, N.; MacKerell, A. D., Jr. J. Comput. Chem. 2000, 21,
(15) Lii, J. H. J. Phys. Chem. A 2002, 106, 8667.
(16) Esposti, A. D.; Alonso, J. L.; Cervellati, R.; Lister, D. G.; Lopez,
(54) Jorgensen, W. L.; Maxwell, D. S.; Tirado-Rives, J. J. Am. Chem.
J. C.; Palmieri, P. J. Chem. Soc., Faraday Trans. 1990, 86, 459. Soc. 1996, 117, 11225.
(17) Allinger, N. L.; Schmitz, L. R.; Motoc, I.; Bender, C.; Labanowski,
(55) Maxwell, D. S.; Tirado-Rives, J.; Jorgensen, W. L. J. Comput.
J. K. J. Comput. Chem. 1992, 13 (7), 838. Chem. 1995, 16, 984.
(18) Malon, P.; Mickley, L. J.; Sluis, K. M.; Tam, C. N.; Keiderling, T.
(56) Jorgensen, W. L.; McDonald, N. A. J. Mol. Struct. (THEOCHEM)
A.; Kamath, S.; Uang, J.; Chickos, J. S. J. Phys. Chem. 1992, 96, 10139. 1998, 424, 145.
(19) Bouchoux, G.; Leblanc, D.; Me´, O.; Ya´n˜ez, M. J. Org. Chem. 1997,
(57) McDonald, N. A.; Jorgensen, W. L. J. Phys. Chem. B 1998, 102,
(20) Li, Z.-H.; Wang, W.-N.; Fan, K.-N.; Wong, M. W.; Huang, H.-H.;
(58) Rizzo, R. C.; Jorgensen, W. L. J. Am. Chem. Soc. 1999, 121, 4827.
Huang, W. Chem. Phys. Lett. 1999, 305, 474.
(59) Price, M. L. P.; Ostrovsky, D.; Jorgensen, W. L. J. Comput. Chem.
(21) Durig, J. R.; Coulter, G. L.; Wertz, D. M. J. Mol. Spectrosc. 1968, 2001, 22, 1340.
(60) Sun, H. J. Phys. Chem. B 1998, 102, 7338.
(22) Durig, J. R.; Li, Y. S.; Tong, C. C. J. Mol. Struct. 1973, 18, 269.
(61) Villa, A.; Cosentino, U.; Pitea, D.; Moro, G.; Maiocchi, A. J. Phys.
(23) Legon, A. C. Chem. Commun. 1970, 838. Chem. A 2000, 104, 3421.
(24) McDermott, P. J. Phys. Chem. 1986, 90, 2569.
(62) Norrby, P.-O.; Brandt, P. Coord. Chem. ReV. 2001, 212, 79. 18002 J. Phys. Chem. B, Vol. 108, No. 46, 2004
(63) Foresman, J. B.; Frisch, Æ. Exploring Chemistry with Electronic
(69) Soetens, J. C.; Millot, C.; Maigret, B.; Bako´, J. Mol. Liq. 2001, Structure Methods, 2nd ed.; Gaussian, Inc.: Pittsburgh, PA, 1996.
(64) Singh, U. C.; Kollman, P. A. J. Comput. Chem. 1984, 5, 129.
(70) Du¨nweg, B.; Kremer, K. J. Chem. Phys. 1993, 99, 6983.
(65) Besler, B. H.; Merz, K. M., Jr.; Kollman, P. A. J. Comput. Chem.
(71) Ue, M. J. Electrochem. Soc. 1994, 141, 3336. 1990, 11, 431.
(66) Carlson, H. A.; Nguyen, T. B.; Orozco, M.; Jorgensen, W. L. J.
(72) Berens, P. H.; Wilson, K. R. J. Chem. Phys. 1981, 74 (9), Comput. Chem. 1993, 14 (10), 1240.
(67) Sun, H.; Mumby, S. J.; Maple, J. R.; Hagler, A. T. J. Am. Chem.
(73) Berens, P. H.; White, S. R.; Wilson, K. R. J. Chem. Phys. 1981, Soc. 1994, 116, 2978.
(68) van Duijneveldt, F. B.; van Duijneveldt-van de Rijdt, J. G. C. M.;
(74) Berens, P. H.; Mackay, D. H. J.; White, G. M.; Wilson, K. R. J.
van Lenthe, J. H. Chem. ReV. 1994, 94, 1873. Chem. Phys. 1983, 79 (5), 2375.
Paleo Solution – Episode 123 Hey folks, Robb Wolf here. Greg Everett in the house and what is this, More exciting than episodes 1 through 69. Got back from Columbus, Ohio late Sunday night. I had a couple very interesting plan rides and interesting people on the plane to put it that way. So that was the national championships and the Olympic trials for women, weight lifting. Congratulation
2 4 I M B R E N N P U N K T Sekundäre Pflanzenstoffe bei terminaler Niereninsuffizienz Es gibt eine Reihe von Möglichkeiten, die Gesundheit zu unterstützen. Neben spor tlicher Betätigung (oder wenigstens regelmäßiger Bewegung), Vorsorgeuntersuchungen sowie dem Vermeiden unnötiger Risiken (Rauchen, Alkohol, schädliche Umwelteinflüsse) kommt es in erster Linie auf eine ausgew
 J. Phys. Chem. B 2004, 108, 17992-18002
J. Phys. Chem. B 2004, 108, 17992-18002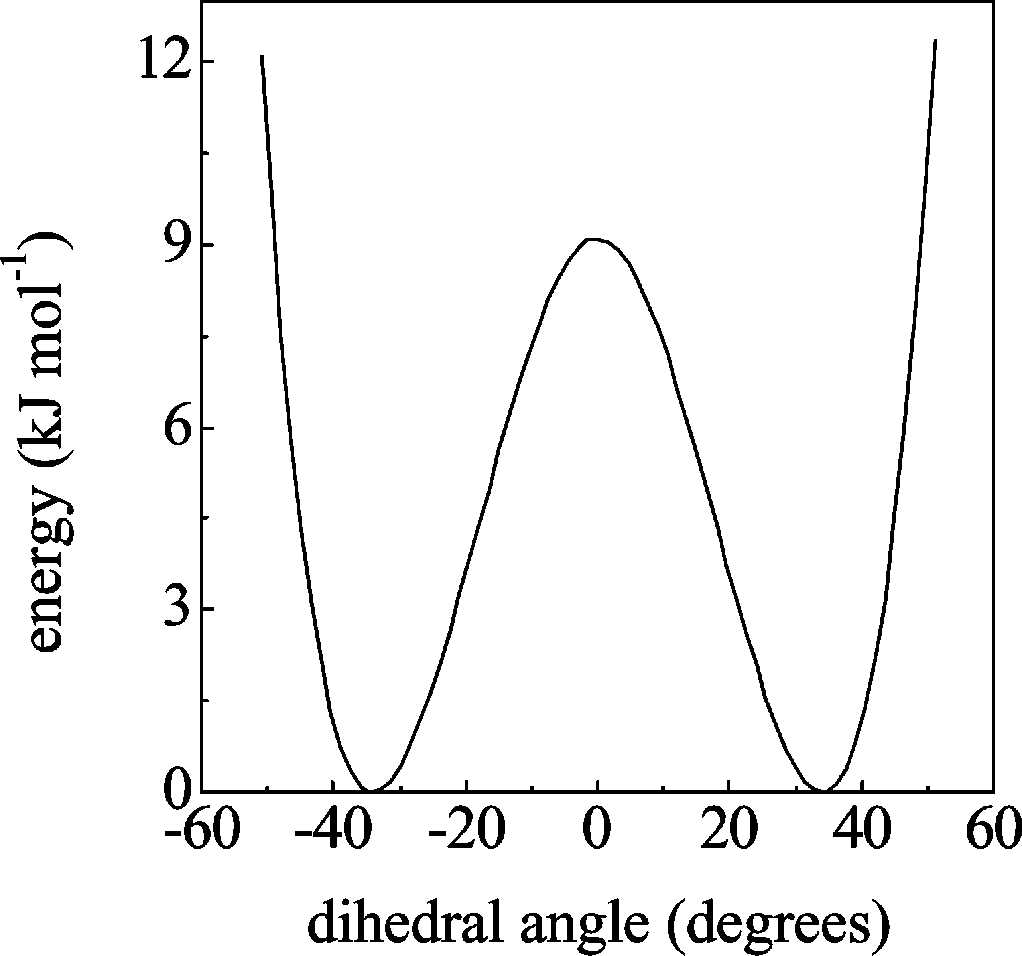
 17994 J. Phys. Chem. B, Vol. 108, No. 46, 2004
17994 J. Phys. Chem. B, Vol. 108, No. 46, 2004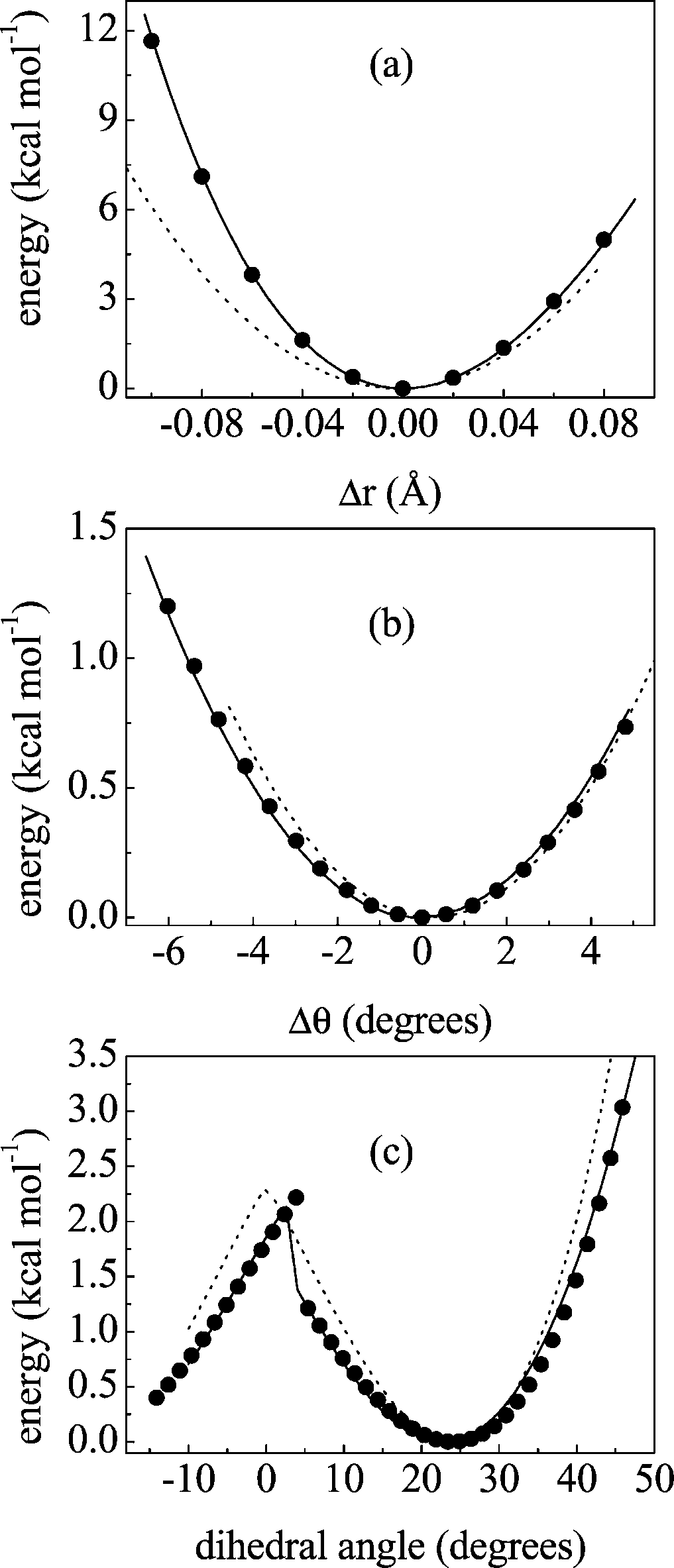
 J. Phys. Chem. B, Vol. 108, No. 46, 2004 17997
J. Phys. Chem. B, Vol. 108, No. 46, 2004 17997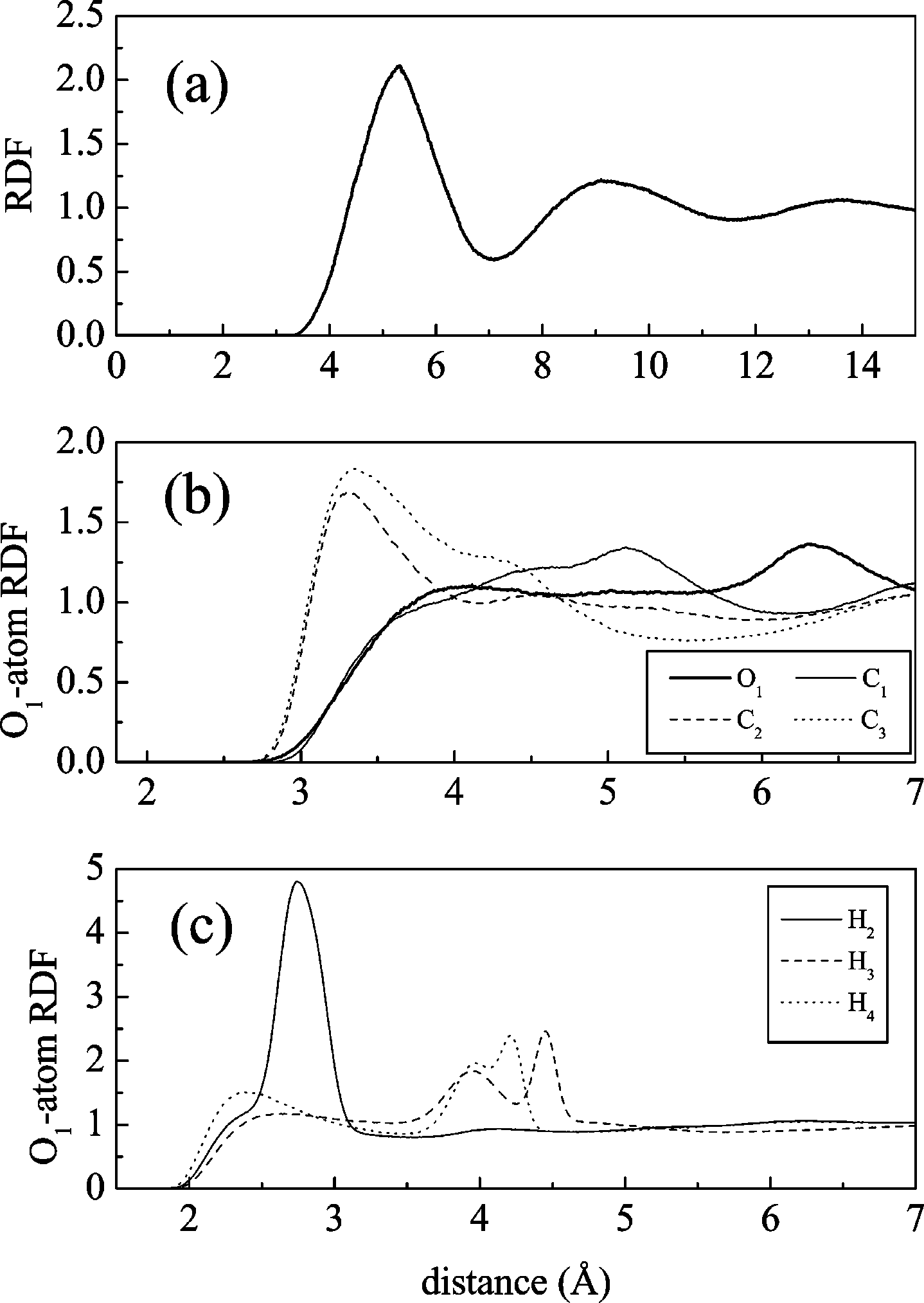
 17998 J. Phys. Chem. B, Vol. 108, No. 46, 2004
17998 J. Phys. Chem. B, Vol. 108, No. 46, 2004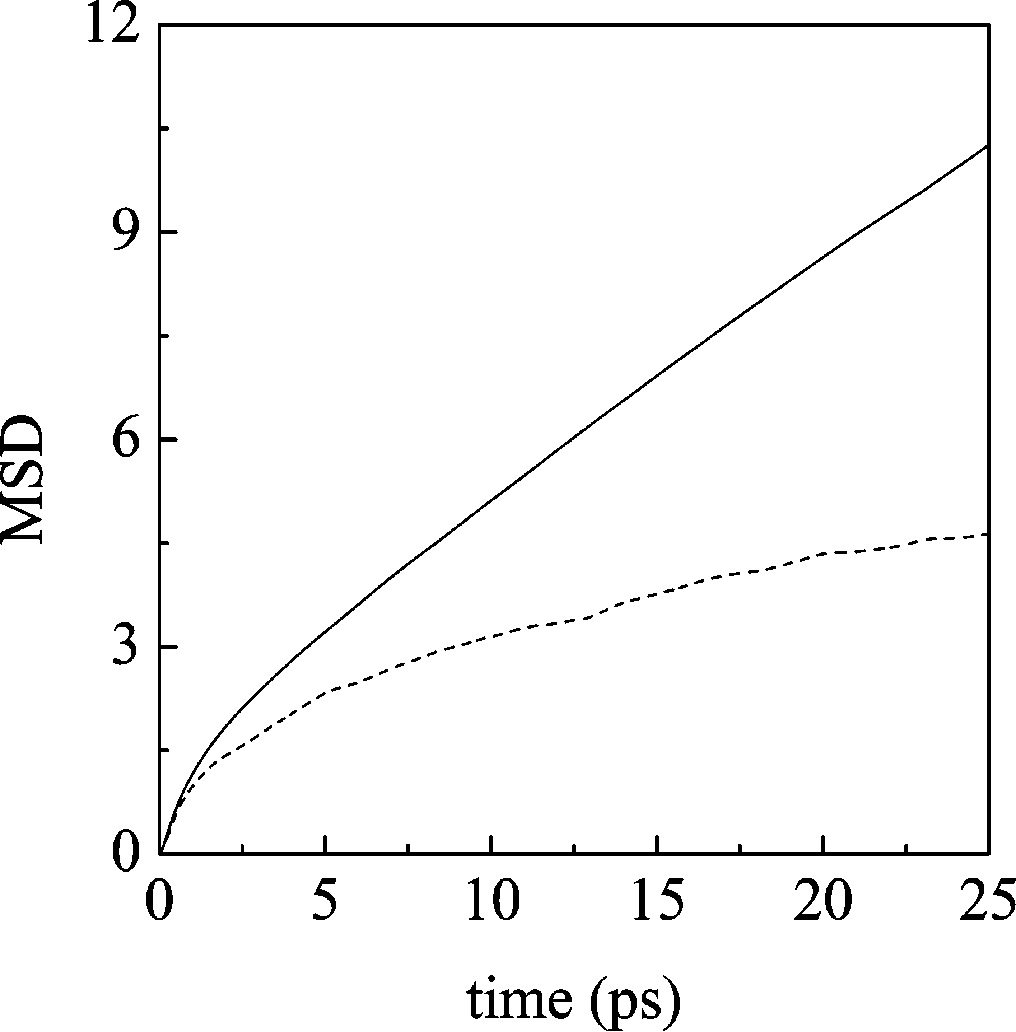
 J. Phys. Chem. B, Vol. 108, No. 46, 2004 17999
J. Phys. Chem. B, Vol. 108, No. 46, 2004 17999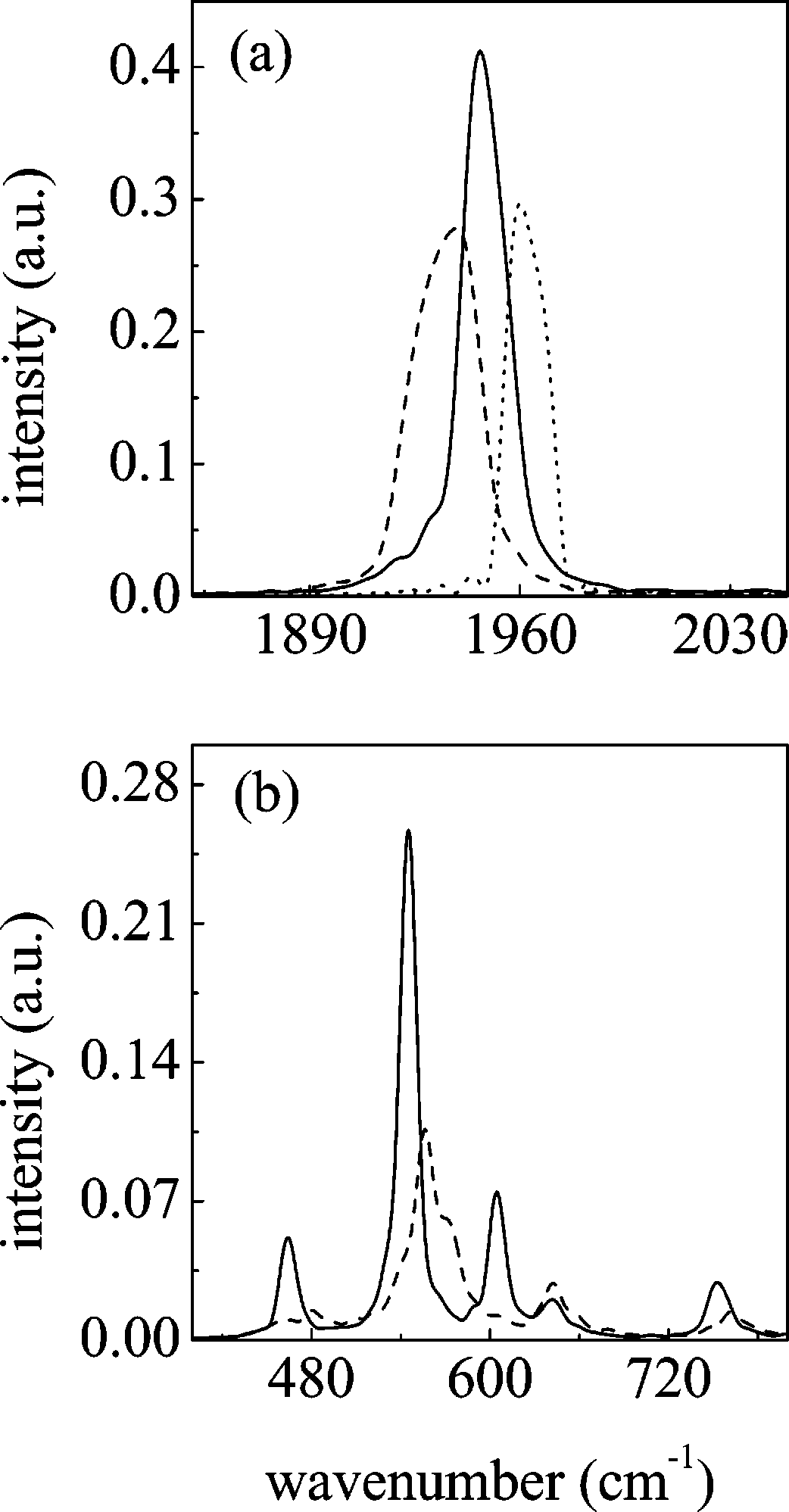 18000 J. Phys. Chem. B, Vol. 108, No. 46, 2004
18000 J. Phys. Chem. B, Vol. 108, No. 46, 2004