Lecture 4 Site-directed mutagenesis
In which we learn how to obtain precise control over the coding content of DNA
The principle of site-directed mutagenesis is that a mismatched oligonucleotide is extended, incorporating the "mutation"into a strand of DNA that can be cloned. In this lecture, I will present a number of current methods in use.
First, let's talk about the approaches in very general terms, because that will allow us to organize the specific methods inour minds. When we talk about making a specific mutation, let's call the molecule that we are starting with, the onewithout the mutation, the "parent" molecule. It might look like this:
The top strand is blue and the bottom strand is magenta. Let's be a bit more specific and point to a specific nucleotide pairthat we intend to change, as follows:
So let us say that in the parent molecule there is a GC base pair that we want to change into an AT base pair. We want themutated version to look like this:
The first general approach is to take the parent molecule and convert it to the mutated version by polymerase chain
reaction. The mutation is made by having a mismatch between the parental template and one (or more) oligonucleotide
primers. You can start with a very small amount of the parent molecule, and by PCR make a tremendous amount of the
mutated version, so much in fact that the chances of cloning the mutated version from the product is essentially 99.999%.
There is always a bit of room for error, of course, so you must carefully confirm your work by DNA sequencing.
To master this approach, we will need to learn a bit more about the design of primers. When you start thinking about
making primers that are slightly mismatched with the template, you have to know what you can get away with and what
Changing the
Let's consider a very simple case, in which you want to make a change in the end of a DNA molecule. end of a PCR
Suppose you have a PCR fragment that looks like this, where the dots indicate an extended sequence
fragment
TCTATGGACCAGTACGATACCAGTA.CGACCTACGTAGACTAGACGGATAGAGAGATACCTGGTCATGCTATGGTCAT.GCTGGATGCATCTGATCTGCCTATCTC
The two oligonucleotides you used to make the fragment look like this:
These oligonucleotides are written 5' to 3'. If you don't understand the oligo on the right, then I suggestthat you go back to review the lecture on PCR.
http://www.escience.ws/b572/L4/L4.htm (1 of 11) [9/7/2002 5:53:32 PM]
Suppose that you want to add an EcoRI site (GAATTC) to the end on the left, and a BamHI site(GGATCC) to the end on the right. Which of these might be correct for the new oligo on the right? 5' GAATTCCTCTATCCGTCTAGTCTA 5' CTCTATCCGTCTAGTCTAGGATCC 5' GGATCCCTCTATCCGTCTAGTCTA 5' ATCTGATCTGCCTATCTCCCATGG
No problem! We just make our two oligonucleotides a bit longer, and embed the extra sequencewithin.
Oligo on the left GCGAATTCTCTATGGACCAGTACGAT
Oligo on the right GCGGATCCCTCTATCCGTCTAGTCTA
The new PCR product would look like this:
GCGAATTCTCTATGGACCAGTACGATACCAGTA.CGACCTACGTAGACTAGACGGATAGAGGGATCCGCCGCTTAAGAGATACCTGGTCATGCTATGGTCAT.GCTGGATGCATCTGATCTGCCTATCTCCCTAGGCG
After we had digested it with the two enzymes, the bits on the ends would be lost and the productwould be ready to clone:
AATTCTCTATGGACCAGTACGATACCAGTA.CGACCTACGTAGACTAGACGGATAGAGGGAGATACCTGGTCATGCTATGGTCAT.GCTGGATGCATCTGATCTGCCTATCTCCCTAG
Please note that we added a "GC" base pair to each end to make the enzymes work better - that is asubject for a future lecture, so don't worry about it just now. The important thing is that we managedto change the ends of the DNA, just by adding a bit of sequence to the 5' ends of each oligonucleotide.
We can do more than append a sequence - we could also change the parental sequence at the endwithout making the product any longer. Remember that our original oligonucleotide pair was:
First, I would like you to note that each is 18 nt in length, and that we would get exactly the sameproduct if we extended one of them at the 3' end, so that it was 21 nt in length.
Why would we get the same product? Because it would look like this, which is the same as ouroriginal version (aside from the coloration)
TCTATGGACCAGTACGATACCAGTA.CGACCTACGTAGACTAGACGGATAGAGAGATACCTGGTCATGCTATGGTCAT.GCTGGATGCATCTGATCTGCCTATCTC
Now let's have some fun, and make a change in the oligo on the left:
http://www.escience.ws/b572/L4/L4.htm (2 of 11) [9/7/2002 5:53:32 PM]
You see that we made a "G" at the third nucleotide instead of a "T". This will create a transversionmutation in the product:
TCGATGGACCAGTACGATACCAGTA.CGACCTACGTAGACTAGACGGATAGAGAGCTACCTGGTCATGCTATGGTCAT.GCTGGATGCATCTGATCTGCCTATCTC
Both strands are affected, because the new version is simply copied into its complementarynucleotides on the bottom strand. So you see, we can make changes in the sequence that are internal.
Why did we extend the oligo on the left so that it was 21 nt in length? Well, we wanted to be sure thatit would anneal correctly to the template in the very first cycle. If we introduce a mismatch, we wantto be sure that there are an adequate number of matching nucleotides at the 3' end of the primer (18 isa safe number, in most cases). So you see that in this mutagenesis approach, the first annealing wouldbe imperfect, and the 5' end of the oligo on the left would be single stranded for three nucleotides.
5'T C G ATGGACCAGTACGATACC----->extensionAGATACCTGGTCATGCTATGGTCAT.GCTGGATGCATCTGATCTGCCTATCTC
Of course, the mutation is copied into the product by extension from the oligo on the right, so once thePCR reaction is underway, the annealing will be perfect over the entire 21 nt of the primer:
5'TCGATGGACCAGTACGATACC----->extension AGCTACCTGGTCATGCTATGGTCAT.GCTGGATGCATCTGATCTGCCTATCTC
So you see, it is fairly straightforward to change a DNA sequence if it can be covered by anoligonucleotide during polymerase chain reaction. Changing the middle of a sequence by two consecutive
Suppose you want to do something a bit more challenging - creating a point mutation in the middle of
reactions;
a DNA sequence, at the position marked with an "*" in the figure:
the 4 oligo method
The ways of doing this in the old days were unspeakable, but now we can simply get on the phone andorder four oligonucleotides; two of which are flanking and two of which cover and introduce themutation into the amplified material:
We perform two PCR reactions to obtain the two halves of our final product, and combine them in athird reaction, using the two "outside" oligonucleotides to generate a chimeric product.
http://www.escience.ws/b572/L4/L4.htm (3 of 11) [9/7/2002 5:53:32 PM]
How does this happen? During the PCR process, the right side of the first molecule can prime thesynthesis from the left side of the second.
Now we can simply cut the PCR product with EcoRI and BamHI, and drop it into the vector, in placeof the original version. Or, we can continue to manipulate the DNA by PCR.
To some extent, this is just using the end-based method we described in the first place, but doing ittwice, and then combining the results into a single product. Inverse PCR
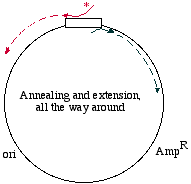
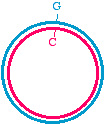
Here's a different approach, which would be appropriate if the DNA template is circular, for examplein a plasmid:
We may start with a circular plasmid, and use two oligonucleotides to change a small region by PCR(see asterisk). The 5' ends of the oligonucleotides are shown not annealed - they do not base pairbecause they are mutagenized. The two oligos are situated in such a way that they re-copy the entireplasmid.
They point towards each other, but only going the "long way around." That is like deciding to go toLos Angeles for the day, but instead of heading down the 405 you go up the Pacific Coast Highway toAlaska, snowshoe over to Denmark, hop a train to Capetown, boat over to Tierra del Fuego, andbicycle up through South and Central America to Los Angeles! It's the long way to communte!
In this PCR example however, it makes sense because it means you don't need to combine two piecesfor cloning. What you obtain in the end is a linear fragment, suitable for reclosure and cloning:
http://www.escience.ws/b572/L4/L4.htm (4 of 11) [9/7/2002 5:53:32 PM]
One comment however, is that the 5' ends of a PCR fragment are exactly the 5' ends of theoligonucleotides. You will need to have a 5' phosphate if you intend to use DNA ligase to reclose thecircle, and so if your oligonucleotide does not have a 5' phosphate (which would be typical) then youneed to apply a phosphate to each end using the enzyme T4 polynucleotide kinase.
One more thing - if you want this to work, you need to use very few template molecules in thereaction, perhaps 1000. If you start with, let's say just to be really gross, a nanogram of template, thenyou will have too much parental DNA lingering by during your transformation. That is, you will findthat very few of your transformed bacteria actually have the mutation in the plasmid. The second general approach to mutagenesis does not use polymerase chain reaction, but does use a polymerase. In
effect, you change only one strand in the parent, and transform the bacteria with the heteroduplex, that might look like
You see that the blue strand has been mutated and is now mismatched with the magenta strand. At this point in the DNA,
the mismatch would make a small bubble of single-stranded DNA. What will the bacteria do with this? When the DNA is
replicated (typically as part of a plasmid), semiconservative replication will cause two different daughter molecules. One
looks just like the parent, and the other has the mutation fixed in both strands:
That's great! Now half of the products will be mutated! Well, that would be true if we could efficiently make the
heteroduplex in the first place (which is a bit of a dubious assumption), and if the heteroduplex could be transformed into
cells with the same high efficiency as the parental version (which may not be true, since it has a bit of single-stranded
DNA in it), and many other worries of a similar nature. We need to have some way of disfavoring the parental version in
this contest. As we are making the mutation in one strand, we need to link that strand to some persuasive form of
That is, we make the mutated (blue) strand a "happy strand" in some way or another, and the parental (magenta) strand
into an "unhappy strand". By the strength of this linkage, we select for the mutated version by disfavoring the parental
When using this approach, it is common to employ single stranded DNA as a template (the magenta strand), because then
you can simply apply a mutagenic oligonucleotide and make a second strand. The second strand you make will not have a
parental strand with which to compete. Think of it as follows:
Start with a double-stranded plasmid with the parental sequence
Then, you make a single-stranded DNA containing just the inner strand of the plasmid, which would look like this:
http://www.escience.ws/b572/L4/L4.htm (5 of 11) [9/7/2002 5:53:32 PM]
Then, apply an oligonucleotide that anneals to the single strand, and carries a mutation. Extend the primer with a DNA
Synthesize a second strand, incorporating the mutagenic primer
Note that there is an A/C mismatch at the top of the figure. Once extension is complete, it is double-stranded and might
look like this. Of course, the Klenow enzyme does not make the blue strand covalently closed. There will be a "nick"
where the synthesis ends, but don't worry about that - once this is transformed into bacteria, the bacterial host will repair
Heteroduplex intermediate, ready to transform into bacteria
Remember once again, that the mutagenized strand will be copied into half of the daughter molecules during replication.
So, that's just terrific, and I hear you wondering how we managed to get the single stranded template with which we
started the method. It is a minor digression. Origins of ssDNA replication - how to get single stranded DNA
As we learned previously, an origin of DNA replication is a required element for ensuring
in the laboratory.
plasmid maintenance. Origins of replication do come however, in several different "colors andstyles." Most commercial plasmids are based on the ColE1 origin, a natural "high copy numberorigin" which fosters the accumulation of several hundred copies of a plasmid per bacterium. Itis also not uncommon to find a second conditional origin of replication in some plasmids,derived from a filamentous bacteriophage such as M13, fd, or f1. These origins of replicationhave two important features:
1. They generate a single-stranded DNA product
They are only activated during co-infection with a helper phage
Why would we want to make single-stranded DNA? One reason would be to make a singlestranded template for a sequencing reaction (a matter we will discuss later in the course), or asingle stranded DNA probe. Site directed mutagenesis is sometimes facilitated by having asingle-stranded plasmid to work with.
In any case, a slight digression to discuss the life-cycle of the filamentous bacteriophage might
http://www.escience.ws/b572/L4/L4.htm (6 of 11) [9/7/2002 5:53:32 PM]
Lifestyles of the filamentous bacteriophage 1. Phage enters through pili of male (F+/Hfr) bacteria 2. Introduced phage genome is single-stranded 3. A double-stranded "replicative form (RF)" is generated 4. New single-stranded genomes are copied from the RF
The new phage carrying the single-stranded genome buds
Those poor male bacteria! They have to contend with invading filamentous phage - somethingthat Rogaine just can't cure! We'll be talking more about male and female bacteria in a laterlecture ("sex" in bacteria isn't quite the same concept as in eukaryotes).
What is significant here is that the virion of the filamentous phage (i.e. the viral particle) carries
a single-strand of DNA - not a double helix. In the cell, this single-stranded genome (2.) is used
as a template to synthesize a double-stranded replicative form (RF), which is essentially a
plasmid (3.). The replicative form is used as a template to generate new single-stranded genomes
(4.) that are packaged into virions (5.) to generate new phage. The cell doesn't die - it just grows
more slowly and continues to secrete phage indefinitely. The practical side of this story - if you use a cloning vector that is based on a filamentous
bacteriophage (such as M13mp18 which is an engineered version of the phage M13) or merely
contains an origin of replication from a filamentous bacteriophage (such as f1), then you can
induce single-stranded DNA replication and collect the products in the form of secreted phage
particles (which may be precipitated from the growth medium with polyethylene glycol). In the
case of a plasmid that only contains an f1 origin of replication, and not the remaining genes from
the phage, it is necessary to infect the plasmid containing cell with a filamentous "helper phage"
that will activate the f1 origin of replication in the plasmid and foster viral secretion.
Now we see how to get single stranded DNA, but what about those persuasive selection methods? The smiley faces and
Fundamentally, we're talking about approaches that allow us to distinguish one strand from another. Restriction
Suppose you had a parental DNA that had a unique
enzymes used restriction site in it. If you mutagenized the restriction site to distinguish at the same time that you made a mutation in your gene of strands
interest, then the parental strand would be sensitive to theenzyme and the other strand containing the mutation wouldnot.
, called theTransformer Site-Directed Mutagenesis Kit. Digestion ofthe heteroduplex with the restriction enzyme debilitates theparental strand, because it introduces a "nick". The DNA
http://www.escience.ws/b572/L4/L4.htm (7 of 11) [9/7/2002 5:53:32 PM]
can then be transformed into a bacterial strain. Theefficiency can be increased by extracting the pooled DNAfrom these cells and digesting a second time. This willeliminate the products of replication in the bacteria that arepurely parental (homoduplex), and will spare the ones thatare purely mutagenized (homoduplex). These plasmids canthen be reintroduced into bacteria, and most of thesurviving plasmids should be the mutagenized form.
Let me give you another example. There is a restrictionenzyme named Dpn I that will cleave the sequenceGMeATC where MeA means that the adenylate nucleotideis methylated. Dpn I will not cleave the unmethylatedsequence GATC. We can methylate such sequences in aplasmid by growing the plasmid in a "dam+" strain ofbacteria. Suppose then that we prepare a single strandedDNA template in such a "dam+" strain. The parental strandwould be methylated at every GATC sequence (that is,approximately every 200 to 300 nt). When we apply anoligonucleotide primer to this template and extend it usingKlenow fragment, however, the new DNA that issynthesized in vitro will be unmethylated. We thereforecreate a marked difference between the parental strand(methylated) and the mutagenized strand (unmethylated).
Once we have completed synthesis of the mutagenizedstrand, what would happen if we tried to digest theheteroduplex with Dpn I?
The answer is that the parental strand would be nicked(cleaved) in many places, but the mutagenized strandwould not. By putting this extra damage into the parentalstrand, it is less favored during replication in the bacteria. Uracil
Here's another method, and this one involves taking
N-glycosylase advantage of the enzyme that we discussed in the first used to
lecture that removes uridylate nucleotides from DNA
distinguish strands
How do we get a parental DNA that contains numerousuracil bases incorporated in place of thymidine bases? Theanswer is that we grow the plasmid in a strain that makesdeoxyuridine triphosphate (a strain that is "dut-", meaningdUTPase deficient) and does not surveil the DNA for uracilto excise (a strain that is "ung-", meaning uracilN-glycosylase deficient). The bacteriologists usually don'tsay "minus" and "plus" by the way - they would just callsuch a strain "dut, ung", meaning that those two loci had
http://www.escience.ws/b572/L4/L4.htm (8 of 11) [9/7/2002 5:53:32 PM]
So we make the single stranded parental DNA in a dut, ung
strain of bacteria, apply the mutagenic oligonucleotide in
vitro, and extend it using the usual DNA substrates and
Klenow fragment. The newly synthesized DNA will not
contain uracil bases, because we did not use dUTP as one
of the substrates - only dATP, dGTP, dCTP, and dTTP.
What would happen if we treated this DNA with uracil
N-glycosylase? Well, the parental strand would be
shredded and the mutagenized strand would be untouched.
We don't actually have to add the enzyme ourselves - we
could just take the heteroduplex and transform a wild type
bacterium with it - one that was not dut, ung that is. The
wild type bacteria would shred the parental strand
specifically, because its uracil N-glycosylase would find
The
"Altered States" - sounds like something that ought to havebeen invented by Kary Mullis, but
method.
1. Start with a plasmid carrying a defective selectable
Link the mutation you are making elsewhere in the
plasmid to a correction of the defective selectablemarker.
Select for correction of the selectable marker, and you
are likely to also find plasmids with your specificmutation introduced as well
Sound's easy? Here's a diagram that help to explain it.
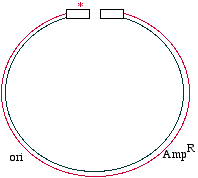
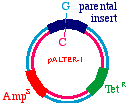
Start with a double stranded plasmid containing your DNAinsert, the sequence you wish to mutagenize:
Note that there is a G/C base pair that we want tomutagenize to an A/T base pair, in our dark blue sequence(the parental insert). Also, there is a green sequencerepresenting tetracycline resistance, and a red sequence thatis a defective version of the ampicillin resistance gene. Since the Amp gene is defective, we will say that it isAmpS meaning "sensitive".
http://www.escience.ws/b572/L4/L4.htm (9 of 11) [9/7/2002 5:53:32 PM]
Now we prepare a single stranded version of the plasmid,perhaps by simply denaturing them in alkali. We annealTHREE oligonucleotides to the circle: One to the DNAparental insert, that causes the mutation in our gene ofinterest (from a G to an A in this example), one to thetetracycline resistance gene that will debilitate it by theintroduction of a mutation, and one to the ampicillin"sensitive" gene that will repair it by the introduction of amutation.
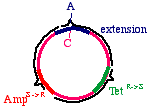
These oligonucleotides are extended clockwise around theplasmid using DNA polymerase Klenow fragment, so itlooks like this:
Note that this heteroduplex has three points of mismatch, inthree entirely different places in the plasmid. The "innerstrand" that contains the parental sequence is unchanged,but the outer strand will contain the three alterations. Whathappens when the bacteria replicates this?
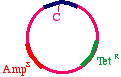
The answer is that two types of products will appear. First,the replicative products of the inner strand:
These will carry an intact tetracycline resistance gene and anonfunctional ampicillin resistance gene. The cells thatinherit these plasmids will die in ampicillin. On the otherhand, the products of the outer strand:
These will carry a functional ampicillin resistance gene, anonfunctional tetracycline resistance gene, and moreimportantly, the G->A mutation in the DNA insert.
So, by transforming the synthetic product into E. coli and
http://www.escience.ws/b572/L4/L4.htm (10 of 11) [9/7/2002 5:53:33 PM]
growing on ampicillin, we favor the mutated strand. the beta galactosidase gene
As you see, when the bacterial streaks are plated inampicillin and the colorimetric substrate X-gal (panel onleft), you get most showing blue color indicating repair ofthe gene. This is an indication of good concordancebetween ampicillin resistance and beta galactosidase genemutation (repair). You also see that very few aretetracycline resistant (panel on right).
Now why should we want to debilitate the tetracyclinegene? So that we can use the method to make additionalchanges, and while we're doing that we will repair thetetracycline gene and debilitate the ampicillin gene. That is,we can make a sequence of changes in our insertion,toggling between ampicillin resistance and tetracyclineresistance. A couple of interesting Michael P. Weiner, Tim Gackstetter, Gina L. Costa, John C. Bauer, and things to read:
Keith A. Kretz (From: Molecular Biology: Current Innovations and Future Trends. Eds. A.M. Griffin and H.G.Griffin. California State University Northridge
1996, 1997, 1998, 1999, 2000, 2001, 2002
http://www.escience.ws/b572/L4/L4.htm (11 of 11) [9/7/2002 5:53:33 PM]
PATIENT NAME: ____________________________________________________________________ WHAT IS THE MAIN REASON FOR YOUR CHILD’S VISIT TODAY______________________________ HOW LONG HAS THIS PROBLEM EXISTED________________________________________________ PLEASE MAKE A CHECK MARK BY YOUR CONCERNS EAR PROBLEMS NOSE PROBLEMS THROAT/MOUTH/NECK PROBLEMS ____HOARSENESS/VOICE

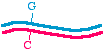
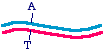 Lecture 4
Lecture 4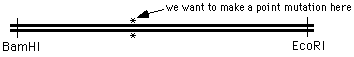
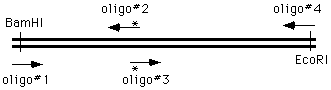 You see that we made a "G" at the third nucleotide instead of a "T". This will create a transversionmutation in the product:
TCGATGGACCAGTACGATACCAGTA.CGACCTACGTAGACTAGACGGATAGAGAGCTACCTGGTCATGCTATGGTCAT.GCTGGATGCATCTGATCTGCCTATCTC
Both strands are affected, because the new version is simply copied into its complementarynucleotides on the bottom strand. So you see, we can make changes in the sequence that are internal.
You see that we made a "G" at the third nucleotide instead of a "T". This will create a transversionmutation in the product:
TCGATGGACCAGTACGATACCAGTA.CGACCTACGTAGACTAGACGGATAGAGAGCTACCTGGTCATGCTATGGTCAT.GCTGGATGCATCTGATCTGCCTATCTC
Both strands are affected, because the new version is simply copied into its complementarynucleotides on the bottom strand. So you see, we can make changes in the sequence that are internal.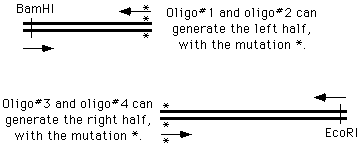

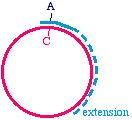
 How does this happen? During the PCR process, the right side of the first molecule can prime thesynthesis from the left side of the second.
How does this happen? During the PCR process, the right side of the first molecule can prime thesynthesis from the left side of the second.
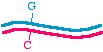
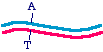

 One comment however, is that the 5' ends of a PCR fragment are exactly the 5' ends of theoligonucleotides. You will need to have a 5' phosphate if you intend to use DNA ligase to reclose thecircle, and so if your oligonucleotide does not have a 5' phosphate (which would be typical) then youneed to apply a phosphate to each end using the enzyme T4 polynucleotide kinase.
One comment however, is that the 5' ends of a PCR fragment are exactly the 5' ends of theoligonucleotides. You will need to have a 5' phosphate if you intend to use DNA ligase to reclose thecircle, and so if your oligonucleotide does not have a 5' phosphate (which would be typical) then youneed to apply a phosphate to each end using the enzyme T4 polynucleotide kinase.
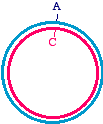 Then, apply an oligonucleotide that anneals to the single strand, and carries a mutation. Extend the primer with a DNA
Synthesize a second strand, incorporating the mutagenic primer
Note that there is an A/C mismatch at the top of the figure. Once extension is complete, it is double-stranded and might
look like this. Of course, the Klenow enzyme does not make the blue strand covalently closed. There will be a "nick"
where the synthesis ends, but don't worry about that - once this is transformed into bacteria, the bacterial host will repair
Heteroduplex intermediate, ready to transform into bacteria
Remember once again, that the mutagenized strand will be copied into half of the daughter molecules during replication.
Then, apply an oligonucleotide that anneals to the single strand, and carries a mutation. Extend the primer with a DNA
Synthesize a second strand, incorporating the mutagenic primer
Note that there is an A/C mismatch at the top of the figure. Once extension is complete, it is double-stranded and might
look like this. Of course, the Klenow enzyme does not make the blue strand covalently closed. There will be a "nick"
where the synthesis ends, but don't worry about that - once this is transformed into bacteria, the bacterial host will repair
Heteroduplex intermediate, ready to transform into bacteria
Remember once again, that the mutagenized strand will be copied into half of the daughter molecules during replication.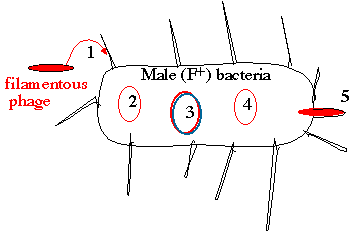 Lifestyles of the filamentous bacteriophage
Lifestyles of the filamentous bacteriophage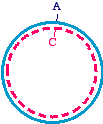 can then be transformed into a bacterial strain. Theefficiency can be increased by extracting the pooled DNAfrom these cells and digesting a second time. This willeliminate the products of replication in the bacteria that arepurely parental (homoduplex), and will spare the ones thatare purely mutagenized (homoduplex). These plasmids canthen be reintroduced into bacteria, and most of thesurviving plasmids should be the mutagenized form.
can then be transformed into a bacterial strain. Theefficiency can be increased by extracting the pooled DNAfrom these cells and digesting a second time. This willeliminate the products of replication in the bacteria that arepurely parental (homoduplex), and will spare the ones thatare purely mutagenized (homoduplex). These plasmids canthen be reintroduced into bacteria, and most of thesurviving plasmids should be the mutagenized form.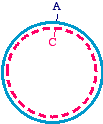
 So we make the single stranded parental DNA in a dut, ung
strain of bacteria, apply the mutagenic oligonucleotide in
vitro, and extend it using the usual DNA substrates and
Klenow fragment. The newly synthesized DNA will not
contain uracil bases, because we did not use dUTP as one
of the substrates - only dATP, dGTP, dCTP, and dTTP.
So we make the single stranded parental DNA in a dut, ung
strain of bacteria, apply the mutagenic oligonucleotide in
vitro, and extend it using the usual DNA substrates and
Klenow fragment. The newly synthesized DNA will not
contain uracil bases, because we did not use dUTP as one
of the substrates - only dATP, dGTP, dCTP, and dTTP.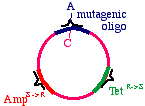
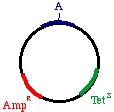 Now we prepare a single stranded version of the plasmid,perhaps by simply denaturing them in alkali. We annealTHREE oligonucleotides to the circle: One to the DNAparental insert, that causes the mutation in our gene ofinterest (from a G to an A in this example), one to thetetracycline resistance gene that will debilitate it by theintroduction of a mutation, and one to the ampicillin"sensitive" gene that will repair it by the introduction of amutation.
Now we prepare a single stranded version of the plasmid,perhaps by simply denaturing them in alkali. We annealTHREE oligonucleotides to the circle: One to the DNAparental insert, that causes the mutation in our gene ofinterest (from a G to an A in this example), one to thetetracycline resistance gene that will debilitate it by theintroduction of a mutation, and one to the ampicillin"sensitive" gene that will repair it by the introduction of amutation. growing on ampicillin, we favor the mutated strand. the beta galactosidase gene
As you see, when the bacterial streaks are plated inampicillin and the colorimetric substrate X-gal (panel onleft), you get most showing blue color indicating repair ofthe gene. This is an indication of good concordancebetween ampicillin resistance and beta galactosidase genemutation (repair). You also see that very few aretetracycline resistant (panel on right).
growing on ampicillin, we favor the mutated strand. the beta galactosidase gene
As you see, when the bacterial streaks are plated inampicillin and the colorimetric substrate X-gal (panel onleft), you get most showing blue color indicating repair ofthe gene. This is an indication of good concordancebetween ampicillin resistance and beta galactosidase genemutation (repair). You also see that very few aretetracycline resistant (panel on right).